Pitfalls in Proteomics: Avoiding Problems That Can Occur Before Data Acquisition Begins (EU)
Some applications of liquid chromatography (LC) involve highly specific “tips and tricks” that are acquired by analysts over time, and they can make the difference between producing data that is either terrible or great. The analysis of peptides by LC coupled with mass spectrometry (LC–MS) is one such area where it can appear that highly skilled analysts possess superpowers or “golden hands” that enable them to acquire better data than anyone else in a laboratory. In the end though, careful attention to detail and a thorough understanding of the conditions and chemistry involved in these analyses can help avoid problems before they occur or accelerate troubleshooting problems when they do appear. For this month’s instalment of LC Troubleshooting, I invited Daniel Meston to share his short list of knowledge, tips, tricks, and advice related to analyzing peptides by LC–MS. There are many details that are important to consider in the preparation of samples and in the instrument prior to LC–MS data acquisition that can increase the likelihood of acquiring high quality data.
- Dwight Stoll
LC–MS has become the gold standard methodology used for proteomics, which is the identification and quantification of protein abundances in biological samples. This is because of the ability of LC–MS to provide sensitive detection for untargeted peptides and identify unknown peptides through tandem MS (MS/MS) fragmentation and comparison of the experimental spectra to theoretical spectra prepared in silico. Subsequently, LC–MS-based proteomics tools have become invaluable in diverse disciplines ranging from immunology to microbiology and food chemistry. However, analysis of biomolecules is generally susceptible to a number of unique and perilous sample preparation pitfalls that can severely degrade the quality of data acquired, even when the most sophisticated LC–MS systems are used, which is because of the unique chemistry and size of large biological macromolecules, such as proteins, peptides, and oligonucleotides to name a few. In this instalment, we provide our views on some of the most common pitfalls in sample and instrument preparation relevant to proteomic analyses by LC–MS. We hope that increasing awareness of these potential problems can help users mitigate them and produce higher quality data in general.
Adding Contamination
All mass spectrometric methods require the ionization of the analytes of interest before they can be separated and ultimately detected. For the peptides encountered in proteomic workflows, the ionization step is generally straightforward because conventional tryptic digestion of proteins often results in amino acid chains with terminal amino groups. Indeed, the protonation of peptides is so favourable that they are often observed as multiply charged positive ions. Given the ease of ionization of peptides, MS can almost be regarded as a “universal detector” for peptide analysis. However, this sensitivity can also be a double-edged sword in the sense that the detector will also respond to very low concentrations of easy-to-ionize contaminants in our samples, thus degrading the overall quality of the acquired data.
Polymers: Polymers are perhaps the most frequently encountered type of contamination in proteomic samples, probably because they are present in most laboratories at levels high enough to matter to LC–MS work. A few examples of polymer sources include skin creams and moisturizing products, pipette tips, chemical wipes that contain an abundance of different polyethylene glycols (PEGs), and siliconized surfaces that contain polysiloxanes (PSs). The presence of these contaminants can be readily recognized in MS spectra by their characteristically large numbers of regularly spaced peaks in the spectra (44 Da spacing for PEG, 77 Da spacing for PS).
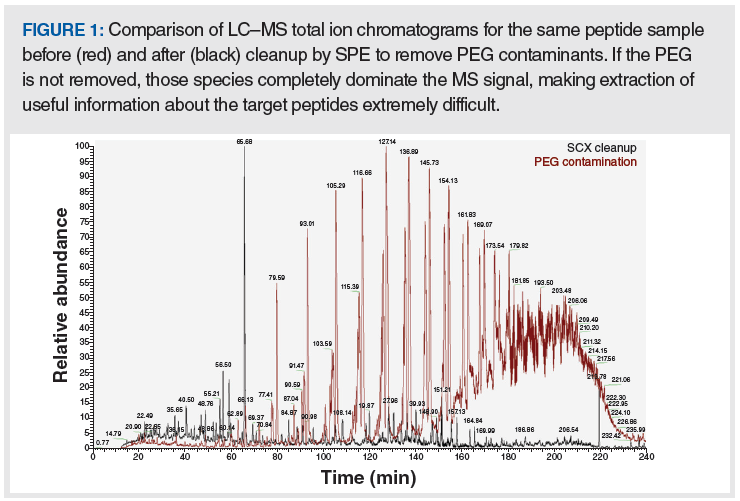
Another source of PEG contaminants is the use of surfactant-based cell lysis methods that are routinely employed in most molecular biology laboratories and involve surfactants such as Tween, Nonident P-40, and Triton X-100. Residual surfactant in samples that are produced using these chemical lysis methods have the potential to produce MS signals that can largely obscure the MS signal of the target peptides, thus rendering the data useless. A dramatic example of such a result is shown in Figure 1. When such protocols are used, extreme care should be taken to selectively remove the surfactants from the sample prior to analysis. Although it is certainly possible to remove PEG contaminants using solid-phase extraction (SPE), it is far easier in the long run to avoid the problem altogether and not use the surfactants for cell lysis in the first place.


Urea: In addition to problems with surfactants, urea used in cell lysis buffers can also cause problems. It is known that urea can decompose to isocyanic acid, which can in turn covalently modify free amine groups in peptides through carbamylation reactions (1). This chemical modification of the peptide can be accounted for in peptide identification software, but only if the software is instructed to look for this.
Residual Salts: In general, residual salts can negatively impact chromatographic performance as well, and they can cause physical damage to fluidics and the MS‑interface instrumentation by scratching the surfaces and clogging the emitter. As a result, residual salts should be removed from the sample prior to injection. Both urea and salts are most commonly removed using a reversed‑phase clean-up step, such as SPE.
Water Quality: Not all high quality laboratory water is created equally, and there are many ways problems can arise with the water used for sample and mobile phase preparation. For example, in-line filters that are used to filter out deoxyribonucleic acid (DNA) can inadvertently lead to contamination of the water with PEG. Even the highest quality water produced on-site in the laboratory can begin to accumulate contaminants within a few days of production. Generally, we should avoid using water that has been sitting around for more than a few days after opening the bottle (if purchased) or production by a polishing system on-site. It is good practice to dedicate a specific subset of mobile phase bottles in the laboratory for LC–MS use only and to avoid washing them with any kind of detergent (2,3).
Keratins: The most abundant protein contamination found in proteomic samples are the keratin proteins that make up our skin, hair, and fingernails. It is not uncommon to observe more than 25% of the peptide content of a proteomics sample to originate from keratin-derived peptides. Any steps that can be taken to reduce this type of contamination will improve the ability to detect low-abundant proteins of interest. In general, natural fibre clothing, such as wool, should not be worn in proteomics laboratories. Ideally, all sample preparation steps should be performed in a laminar flow hood to prevent dust and skin from the analyst and laboratory air from entering samples. Additionally, gloves should be worn at all times and replaced after touching a contaminated surface, such as stopwatches, pens, and laboratory notebooks. However, once the proteins have been digested, one should consider not wearing gloves because these can be a further source of polymer contamination.
Protein and Peptide Adsorption
Peptide adsorption to the LC columns has been addressed in previous instalments of “LC Troubleshooting” (4). A lesser appreciated pitfall in proteomics sample preparation is adsorption of the peptides of interest to the vessel used for sample pretreatment (for example, digestion with trypsin), as well as LC sample vials. Peptides are multifunctional molecules with tremendous diversity in properties, including hydrophobicity and the number of acidic and basic functional groups. These properties make them prone to adsorption onto materials with very different properties, such as glass and plastics. This observation is also true for proteins but to an even greater degree (5). Adsorption of peptides from analytical samples onto such surfaces has been observed within timeframes as low as an hour after being placed in the LC vial, and they can result in a significant decrease in the apparent concentration of low-abundant peptides. A number of strategies to minimize sample adsorption to glass LC vials have been demonstrated, including the use of surfactants (which is not recommended for LC–MS, as discussed above). “Priming” vessels with a sacrificial protein, such as bovine serum albumin (BSA), is also done, similar to what is done with LC columns (4). The idea here is that the vessel is rinsed with a solution of a protein that is unrelated to the target proteins being analyzed (or synthetic peptides not found in biological samples); adsorption sites on the material are subsequently saturated and therefore unable to further adsorb peptides or proteins of interest from the analytical sample. Many vendors also offer “high-recovery” LC vials and other products that are engineered to minimize such undesirable analyte adsorption. An additional point to be aware of in this area is related to the removal of the sample solvent during the sample preparation process (for example, vacuum centrifugation to remove organic solvents). Completely removing all of the solvent promotes strong analyte adsorption onto surfaces; thus, it is helpful to avoid complete drying and to leave a small amount of liquid in the vial to increase analyte recovery.
Plastic micropipette tips present another opportunity to lose peptides to adsorption. With this in mind, it is important to limit the number of sample transfers during sample preparation, which has spurred the development of a number of so-called “one-pot” sample preparation methods that minimize contact between the sample and vessel, including nanoPOTS (6), SP3 (7), and FASP (8). From this research, it is becoming increasingly clear that single reactor vessel sample preparation protocols are superior to conventional protocols in the proteomics area.
Finally, when transferring peptide samples, it is best to avoid contact with metals. For example, most glass syringes are fitted with stainless steel needles by default. If a peptide sample is drawn up through a metal needle, peptides can be lost to adsorption on the metal surface. One way to avoid this scenario when using glass syringes is to remove the plunger, fill the sample into the glass syringe barrel using a glass pipette, and then push the sample out of the syringe using the plunger, but through a PEEK capillary fitted to the syringe via a Luer lock fitting, rather than pushing it through the metal needle. Doing so is particularly important in the case of MS calibrations where many vendors utilize a peptide calibrant that can be depleted by metal syringe needles.
Sample Matrix Incompatibilities
Other aspects of the sample matrix are worthy of careful consideration. Although it is attractive to consider using trifluoroacetic acid (TFA) as a mobile-phase additive to improve chromatographic performance for peptides (that is, better peak shape, and increased retention, especially for hydrophilic peptides), this practice is generally frowned upon in the proteomics community because TFA can dramatically suppress peptide ionization, leading to lower overall detection sensitivity compared with using formic acid. An alternative to using TFA in the mobile phase is to add it to the sample and use formic acid to acidify the mobile phase. In this case, the TFA can enhance the retention of hydrophilic peptides on the precolumn, preventing them from eluting into the waste line. Meanwhile, the TFA exiting the precolumn is diverted to waste rather than entering the analytical column and MS interface.
Proteomics applications, which are outside conventional peptide sequencing, may also require specific buffers that preserve the structure or stability of proteins during analysis. It is not uncommon for these buffers to lead to salt precipitation on the electrospray emitter and the source cone of a MS system. In this way, one must optimize the conditions to use volatile buffers, such as ammonium acetate, or to physically clean the surfaces with water to prevent significant sensitivity loss. Recently, it has been demonstrated that postcolumn suppressor technology provides more flexibility in terms of the types of buffers that can be used, some of which are normally considered MS-incompatible (9).
Finally, the age of prepared mobile phases can be particularly problematic in proteomics applications where nano-flow systems are prevalent and mobile phase consumption is so low that a 100-mL batch of the mobile phase can last several weeks. Specifically, when buffer components with differing volatilities are mixed together, there can be selective evaporation of the more volatile component that will change the elution or ionic strength of the mobile phase, which can lead to changes in both absolute and relative peptide retention times. Primarily, this occurrence comes from incorrect viscosity values that change the mixing behaviour of modern nanoLC instruments. Additionally, changes in ionic strength will have a direct effect on the retention factor of the analytes, which will lead to variation in retention time. As a rule of thumb, mobile phases should be replaced every 1–2 weeks to ensure reproducible results. Additionally, in the case of pumping systems that rely on known mobile phase viscosities for accurate flow rates, the mobile phase viscosity should be determined when changing solvents. Instruments that require this feature build in software routines to facilitate these measurements. An additional point is to consider not using conventional membrane degassers when using nano-flow rate because the flow rate is so slow during this process. It has been noted in our laboratory that volatile buffer components, such as formic acid, are preferentially removed from the mobile phase during degassing. In these situations, it is better to utilize alternative degassing measures, such as sonication, that have a low risk for additional contamination of the mobile phase.
Optimizing the Electrospray Source Voltages in nanoLC–ESI–MS
Finally, as we reach the last step prior to the actual acquisition of LC–MS data, we must think about settings for the MS detector itself. Arguably, one of the most consequential missed opportunities in data acquisition for proteomics workflows is related to the optimization of electrospray source voltages. Once analytes are ionized, they are steered through the MS instrument in the direction of the detector using carefully manipulated electric fields. Therefore, analyte detection is highly dependent on the voltage settings for the ion optics that guide the motion of the ionized analytes. Failure to optimize these voltages can result in peptides over large regions of the mass range not making it to the detector, or significantly reduced numbers of detected ions.
Before analyzing any experimental samples, it is worth taking the time to incrementally change the ion source parameters step-wise (for a representative ion infused at the flow rate that will be used in the LC–MS method, we recommend the doubly charged ion of Glu-Fib or angiotensin II), while monitoring the signal change and the spectrum quality. For most modern MS instruments, this tuning step can be done automatically within the MS data acquisition software, but it can be beneficial to further manually fine-tune parameters to maximize signal for the ions of interest. Finally, once the detector settings have been adjusted, it is a good idea to analyze a well-characterized quality control (QC) sample (for example, a cytochrome C digest or HeLa cell digest) to enable a final check on the data quality before analyzing any novel samples.
Summary
Because of the size and varied chemistry of peptides found in proteomics samples, it is important to pay attention to details of each step leading to the acquisition of LC–MS data. This includes sample preparation chemistries, sample handling, optimization of instrument parameters, and control of mobile phases. Paying attention to these details from the start can help avoid data quality problems entirely and simplify troubleshooting when problems do arise. We hope that the tips and tricks discussed here are food for thought for analysts of all levels of experience, and ultimately improve data quality and minimize loss of precious samples and instrument time because of unanticipated problems.
References
1) S. Sun, J.-Y. Zhou, W. Yang, and H. Zhang, Anal. Biochem. 446, 76–81 (2014). https://doi.org/10.1016/j.ab.2013.10.024
2) D.R. Stoll and T. Taylor, LCGC Europe 33(6), 284–288 (2020).
3) D.R. Stoll and T. Taylor, LCGC Europe 33(7), 336–340 (2020).
4) D.R. Stoll, LCGC North Am. 40, 471–475, 483 (2022).
5) Y. Van Wanseele, K. Maes, K. Lanckmans, et al., Chromatographia 81, 65–72 (2018). https://doi.org/10.1007/s10337-017-3397-9
6) J. Woo, S.M. Williams, L.M. Markillie, et al., Nat. Commun. 12, 6246 (2021). https://doi.org/10.1038/s41467-021-26514-2
7) C.S. Hughes, S. Moggridge, T. Müller, et al., Nat. Protoc. 14, 68–85 (2019). https://doi.org/10.1038/s41596-018-0082-x
8) J.R. Wiśniewski, A. Zougman, N. Nagaraj, and M. Mann, Nat Methods 6, 359–362 (2009). https://doi.org/10.1038/nmeth.1322
9) S. Wouters, I. Pijpers, N. Vanden Haute, et al., Front. Anal. Sci. 2, 1002935 (2022). https://doi.org/10.3389/frans.2022.1002935
ABOUT THE CO-AUTHOR
Daniel Meston is a postdoctoral researcher at the Free University of Brussels in Belgium. He also serves as the vice-president of the British Chromatographic Society.
ABOUT THE COLUMN EDITOR
Dwight R. Stoll is the editor of “LC Troubleshooting”. Stoll is a professor and the co-chair of chemistry at Gustavus Adolphus College in St. Peter, Minnesota, USA. Direct correspondence to: amatheson@mjhlifesciences.com







, Its Unique Inventor James Lovelock (1919–2022), and GAIA (EU)")

")







; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")



New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.


, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

