Hydrophilic Interaction Chromatography
This article details the principles of hydrophilic interaction chromatography (HILIC) and its complementary selectivity to reversed-phase high performance liquid chromatography (HPLC). Advantages of the technique that result from the use of low-viscosity, high-organic concentration mobile phases will be demonstrated. For example, LC–mass spectrometry (MS) sensitivity is enhanced and higher flow rates and longer columns can be used effectively with such mobile phases in HILIC. Common stationary phases employed in HILIC are reviewed.
This article details the principles of hydrophilic interaction chromatography (HILIC) and its complementary selectivity to reversed-phase high performance liquid chromatography (HPLC). Advantages of the technique that result from the use of low-viscosity, high-organic concentration mobile phases will be demonstrated. For example, LC–mass spectrometry (MS) sensitivity is enhanced and higher flow rates and longer columns can be used effectively with such mobile phases in HILIC. Common stationary phases employed in HILIC are reviewed.


Hydrophilic interaction chromatography (HILIC) is a liquid chromatography (LC) technique that uses a polar stationary phase (for example, silica or a polar bonded phase) in conjunction with a mobile phase containing an appreciable quantity of water (usually at least 2.5% by volume) combined with a higher proportion of a less polar solvent (often acetonitrile). Most commonly, separations are carried out using 5–40% water (or aqueous buffers); the technique also is compatible with gradient elution. The term HILIC was coined by Alpert in 1990, who explained its principles and some important applications (1). However, the technique dates back at least to the development of separation of sugars in the 1970s, where aminopropylsilane phases were used typically with around 80% acetonitrile. Even in 1941, Martin and Synge (2) separated amino acids on a silica column using water-saturated chloroform, claiming that the separation mechanism was a partition between the water layer on the silica and chloroform. It was later discovered that an immiscible organic solvent was not a requirement for the separation, as indeed is the case for HILIC. In HILIC, hydrophilic, polar, and charged compounds are retained preferentially compared with hydrophobic neutral compounds — the opposite of reversed-phase LC. For example, toluene is eluted in the void volume in a typical HILIC separation, whereas it shows strong retention in reversed-phase LC using a C18 column. Conversely, uracil, which often is used as a void volume marker in reversed-phase LC, can show good retention in HILIC separations. Increasing the proportion of the aqueous component of the mobile phase in HILIC generally gives reduced retention. The order of solute elution resembles that of normal-phase chromatography and indeed, HILIC sometimes is called "aqueous normal-phase chromatography"; some studies suggest there can be at least some similarity in the separation mechanism of the two techniques. The surface of commonly used silica columns in HILIC is deactivated by the presence of the significant amounts of water in the mobile phase in comparison with regular normal-phase chromatography. As a result, fewer peak shape problems are obtained for strongly polar solutes in comparison to normal-phase separations.
Advantages of HILIC
HILIC can be regarded as a complementary technique to reversed-phase HPLC, owing to the differences in elution order of solutes and its good retention of hydrophilic species, which are difficult to retain in reversed-phase LC. In addition, it has the following advantages:
• Enhanced detection sensitivity when used in conjunction with mass spectrometry (MS). The high organic content of the mobile phase in HILIC allows for efficient spraying and desolvation in electrospray MS; a sensitivity increase of as much as three orders of magnitude was demonstrated for the analysis of the bronchodilator salbutamol in comparison with reversed-phase HPLC–MS (3).
• It might be possible to inject extracts directly from C18 solid-phase extraction (SPE) columns, from which solutes usually are eluted with high organic content. Use of an HPLC mechanism that is different from that of the sample preparation also introduces a degree of orthogonality into the overall procedure.
• Higher flow rates can be used due to the low viscosity of mobile phases of high organic content; the viscosity of 90% acetonitrile at 20 °C is 0.43 cP, about half the 0.90–0.86 cP for 10– 30% acetonitrile, typically used for separation of hydrophilic solutes by reversed-phase HPLC. In addition, solute diffusivity is increased in HILIC mobile phases. Thus, at high flow rate, relatively greater efficiencies can be obtained compared with reversed-phase HPLC as the C term region of the van Deemter plots is flatter (see Figure 1).


Figure 1
• Alternatively, low-viscosity mobile phases allow the use of columns around twice as long as the equivalent reversed-phase columns, while maintaining a similar back pressure. Superficially porous silica phases with small particle size (dp = 2.7 μm, thickness of porous shell 0.5 μm) recently have become commercially available (Halo, Advanced Materials Technology, Wlimington, Delaware). Very high efficiencies with low backpressure have been achieved with C18 particles; this advantage can be combined with that of the low viscosity of HILIC mobile phases. Figure 2 shows the separation of some acids, bases, and neutrals with three coupled 15-cm bare silica Halo columns, in which over 100,000 theoretical plates are generated with a total system backpressure of only 280 bar, which is well within the pressure capability of a conventional HPLC system. The void time of this separation was only 4.4 min, suggesting the possibility of real practical applications.

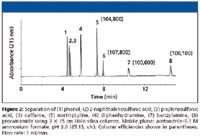
Figure 2
• Peak shapes are generally good for basic compounds, while there can be problems of low efficiency and tailing for these compounds in reversed-phase HPLC.
• Column overload is sometimes cited as a potential weakness of HILIC (4). However, overload of ionized basic compounds actually can be less serious than for reversed-phase HPLC, providing the mobile phase conditions are optimized (see Figure 3) (5). Overloaded peaks in HILIC often show fronting, whereas tailing usually is shown in reversed-phase LC. Fronting can be the result of solute-solute interactions in the stationary phase.


Figure 3
Mechanism of HILIC Separations
The separation mechanisms involved in HILIC are undoubtedly complex, and have been discussed in detail in a recent review (4). Alpert (1), based upon the previously suggested mechanism for the separation of carbohydrates on aminopropylsilane columns, suggested that a stagnant aqueous layer was held in the vicinity of the stationary phase and that partitioning of solutes took place between this layer and the bulk mobile phase. In this case, HILIC would be distinct from normal-phase chromatography, which involves adsorption directly onto the stationary phase. Alpert considered that dipole–dipole interactions might be responsible for the partitioning of solutes between the two layers. However, other authors (6) suggested that the HILIC mechanism was similar to that in normal-phase chromatography, considering that the interaction forces encompassed both dipole–dipole interactions and hydrogen bonding. Plots of the retention factor against the fraction of water in the mobile phase have been used in an effort to establish whether the mechanism involves partitioning or adsorption. In conventional normal-phase systems, retention usually is described by the Snyder–Soczewinski equation:
log k = log kB – n log XB
where n is the number of strong solvent (B) molecules displaced by the solute, kB is the value of k when pure B is used, and XB is the mole-fraction of the stronger B solvent in the mobile phase.
A more approximate form of the equation substitutes the volume fraction of B φ for the mole fraction.
Thus, a plot of log k versus log of the mole fraction of water (the B solvent) or (more approximately) log of the volume fraction of water, should yield a straight line if the mechanism is adsorption.
A plot of log k versus the volume fraction of water should yield a straight line. Hemström and Irgum (4) compared such plots for several sets of published HILIC data, but were unable to reach firm conclusions as to whether the mechanism was principally partition or adsorption. A complication was likely to be the contribution of ionic retention effects, for example from silanol groups on silica-based columns. It is possible that the mechanism can be a mixture of adsorption and partition (and ion exchange for some solutes). Nevertheless, many workers continue to regard partitioning as a major contributor to the separation mechanism.
Alpert (7) recently introduced a variant of HILIC termed electrostatic repulsion hydrophilic interaction chromatography (ERLIC). Here, an ion-exchange column is eluted with a predominantly organic mobile phase. Solutes can be retained by HILIC mechanisms even if they have the same charge as the stationary phase. Depending upon the magnitude of the solute charge, HILIC retention is reduced by repulsion between the charged groups on the solute and stationary phase. Mixtures of peptides, amino acids, and nucleotides, which normally require gradient elution in ion-exchange separations, were separated isocratically (7).
Stationary Phases for HILIC
A range of polar phases is available for HILIC separations including bare silica, aminopropyl, diol, and zwitterionic (for example, sulfoalkylbetaine) phases; bonded phases can be based upon silica or on organic polymer matrices. As is usual in HPLC, polymeric supports offer use of the column over a wider pH range (which might be useful in manipulating selectivity); however, column efficiencies can be lower than with silica-based phases. Silica-based phases represent by far the larger group of columns specifically designed for HILIC. Alpert (1) noted the pronounced retention of solutes containing charged basic groups due to the strong hydrophilicity of such species; on silica-based phases, this retention can be enhanced by ionic retention between ionized silanol groups and bases that are protonated or partially protonated in the mobile phase. Conversely, strongly acidic compounds can experience repulsion from silanols and low retention, but manipulation of mobile phase conditions can improve retention (5).
Bare silica phases remain the most popular (and generally the cheapest!) of all HILIC materials (4). A few underivatized silica phases have been marketed especially for HILIC separations (for example, Atlantis, Waters, Milford, Massachusetts). This phase has been shown to give excellent separations of basic compounds (5). Despite possible repulsion effects between ionized silanols and acidic solutes, Guo and Gaiki (8) showed reasonable retention and separation of substituted salicylic and acetic acids on a bare silica phase. Bare silica is advantageous in HPLC–MS applications, because columns have no organic bonded ligands that might bleed and contribute to detector background. One study showed considerable selectivity and peak shape differences between different bare silicas used in the HILIC mode (9). However, this study incorporated some older "Type A" silicas, which contain large and variable quantities of metal impurities. A more recent study using more modern "Type B" silicas, which are consistently low in metal impurities, showed little difference in selectivity for a range of different solutes (5).
Aminopropyl bonded-phases are used widely for the separation of sugars, for which they show specific advantages. Sugars exist in two anomeric forms that can interconvert slowly at acid and neutral pH, giving two peaks for each solute that can lead to overcrowding in the chromatogram (10). The pH within the pores of an amino phase might be as high as 10, due to the strongly alkaline environment produced by the basic groups. At alkaline pH, interconversion of sugar anomers is rapid, and a single peak is obtained. However, the high pH can cause hydrolysis of the bonded ligand and dissolution of silica-based phases, although the column eventually can reach equilibrium due to the formation of acidic silanol groups. Amino phases based upon polymeric supports therefore can be considered preferable. However, stability problems and also the possible reaction of amino phases with aldehydes and ketones in samples have tended to limit somewhat their use outside of the specialist sugar applications.
Sulfoalkylbetaine phases are marketed by SeQuant (Umea, Sweden, now part of Merck, KGaA) especially for HILIC separations and contain the functional group
~ CH2-N+ (CH3)2-CH2-CH2-CH2-SO3 -
bonded to silica or a polymeric matrix. While being zwitterionic, the ionic groups, separated only by a short alkyl chain, might be too close together to give substantial independent interactions with cationic or anionic solutes; in effect, the column charges are considered to mostly cancel each other out, leaving only weak ionic interactions. The polar nature of these groups is still effective in forming an aqueous layer for HILIC separations. It is not clear whether the underlying matrix of the silica version of this phase also could contribute to ionic effects.
A collection of phases including diol, amide and cyano columns have neutral groups, often bonded to silica. Some authors claim that these phases give improved stability, better reproducibility, reduced activity, and faster equilibration times compared with bare silica.
There are several other types of HILIC phase, details of which can be found elsewhere (4). Guo and Gaiki (8) showed considerable selectivity differences between four silica-based phases, aminopropyl, amide, zwitterionic and bare silica, for the separation of some acidic solutes (see Figure 4). They attributed increased retention and different selectivity on the aminopropyl phase to ionic interactions. The improved resolution of aspirin and 4-aminosalicylic acid, compared with that on a bare silica phase, was suggested to be due to electrostatic interaction between the acids and the negatively charged sulfonate groups on the sulfoalkylbetaine phase (although see also previous text). Figure 5 shows another comparative separation, again showing marked selectivity differences, in this case for water-soluble vitamins using a diol silica, a bare silica, and a zwitterionic silica phase. Figures 4 and 5 show no particular evidence for excess activity of bare silica. It is possible that such problems might be encountered with older, less pure silica phases. Further similar studies should enable more detailed elucidation of the fundamental origins of these selectivity differences between stationary phases.

Figure 4
Mobile Phases for HILIC
For the separation of nonionizable compounds, mixtures of acetonitrile and water are popular. For ionizable compounds, choice of buffer is restricted due to the low solubility of inorganic buffers (for example, the commonly used phosphate in reversed-phase HPLC) in mobile phases that contain high proportions of organic solvents. Acid additives such as formic and acetic acid have been used widely, especially as the stability of silica-based columns is compromised at higher pH. The stability of bare silica can be a problem, because the surface is not shielded by the presence of bonded ligands. The pH of formic and acetic acids is raised in solutions of high organic content, as the dissociation of the acids is suppressed. As a result, the ionic strength of simple acid solutions is low, and this can cause peak shape and overloading problems for ionized solutes (5). In such cases, ammonium formate and ammonium acetate buffers are preferable; these are soluble in high concentrations of acetonitrile, maintain a reasonable ionic strength, and also are volatile for LC–MS applications. To improve the retention of poorly retained solutes, alcohols such as methanol can be substituted for some of the water content, as alcohols have a weaker elution strength.


Figure 5
Gradient elution is applicable in HILIC (for example, see Figure 5). Note that a gradient from a solution of low water, high-organic solvent concentration (weak solvent); to one of increased water, reduced-organic solvent concentration (strong solvent) is utilized in HILIC.
Solute injections should be carried out in the mobile phase or a weaker solvent (that is, containing less water) to prevent loss of efficiency. An alternative for samples not very soluble in high concentrations of organic is to use very small sample volumes to keep effects to a minimum. Nevertheless, a drastic solvent mismatch can produce very high apparent efficiencies under some circumstances, for example, injection of the basic drug nortriptyline dissolved in 25% acetonitrile–aqueous acid with a mobile phase of 90% acetonitrile–aqueous acid buffer gave >650,000 plates (see Figure 6). This phenomenon might be a focusing effect caused by the plug of injected water as it travels through the column, and was observed for a number of ionized bases including nortriptyline, diphenhydramine, and propranolol, which appeared to have appropriate k in the mobile phase (11).


Figure 6
Conclusions
Although not as versatile as reversed-phase LC, HILIC is a complementary separation technique with a different selectivity and considerable advantages for suitable solutes, many of which result from its use of high-organic, low-viscosity mobile phases. High column efficiencies and good column capacity are shown for ionizable compounds and compatibility with MS is a particular advantage. Further selectivity differences result from use of different stationary phases, an option facilitated by the much wider range that is becoming available commercially.
References
(1) A.J. Alpert, J. Chromatogr. 499, 177–196 (1990).
(2) A.J.P Martin and R.L.M. Synge, Biochem. J. 35, 1358–1368 (1941).
(3) E.S. Grumbach, D.M. Wagrowski-Diehl, J.R. Mazzeo, B. Alden, and P.C. Iraneta, LCGC 22, 1010–1023 (2004).
(4) P. Hemström and K. Irgum, J. Sep. Sci. 29, 1784–1821 (2006).
(5) D.V. McCalley, J. Chromatogr., A 1171, 46–55 (2007).
(6) T. Yoshida, J. Biochem. Biophys. Meth. 60, 265–280 (2004).
(7) A.J. Alpert, Anal. Chem. 80, 62–76 (2008).
(8) Y. Guo and S. Gaiki, J. Chromatogr., A 1074, 71–80 (2005).
(9) B.A. Olsen, J. Chromatogr., A 913, 113–122 (2001).
(10) U.D. Neue, HPLC Columns (Wiley-VCH, New York, 1997).
(11) D.V. McCalley, unpublished observations.






















