Rigid Porous Polymer Monoliths as Stationary Phases and Supports
Special Issues
The never-ending quest for separation media that enable efficient high speed/high throughput chromatography has led to the design of stationary phases in monolithic formats with both vastly improved mass transfer properties and reduced discontinuity. Historically, porous polymer monoliths have first emerged in the late 1980s/early 1990s followed by their silica-based counterparts in the mid 1990s. The common denominator for both organic and inorganic monoliths was originally their use in HPLC columns. However, the range of applications of monolithic materials grew significantly since their early times. This short review summarizes information about monoliths produced in different shapes such as discs, tubes, columns, polymer layer open tubes (PLOT capillaries), and microfluidic devices, and presents selected applications including chromatographic separations, sample preparation, and enzyme immobilization.
The never-ending quest for separation media that enable efficient high speed/high throughput chromatography has led to the design of stationary phases in monolithic formats with both vastly improved mass transfer properties and reduced discontinuity. Historically, porous polymer monoliths have first emerged in the late 1980s/early 1990s followed by their silica-based counterparts in the mid 1990s. The common denominator for both organic and inorganic monoliths was originally their use in HPLC columns. However, the range of applications of monolithic materials grew significantly since their early times. This short review summarizes information about monoliths produced in different shapes such as discs, tubes, columns, polymer layer open tubes (PLOT capillaries), and microfluidic devices, and presents selected applications including chromatographic separations, sample preparation, and enzyme immobilization.



Monoliths are separation media in the format that can be compared to a single large "particle" that does not contain interparticular voids. As a result, all the mobile phase must flow through the stationary phase. This convective flow greatly accelerates the rate of mass transfer. In contrast to diffusion, which is the typical driving force for mass transfer within the pores of particulate stationary phases during chromatographic processes, convective flow through the pores enables a substantial increase in the speed of the separation of large molecules such as proteins, nucleic acids, or synthetic polymers. A thorough theoretical treatment of the mass transfer within monolithic materials has been developed by Liapis (1) and Tallarek (2).
Although a few approaches to monolithic stationary phases have been suggested in the past, they were not successful and research concerning the monolithic formats has started only in the late 1980s. The focused work of several groups led to a family of useful monoliths comprising compressed hydrophilic gels, macroporous polymer disks, columns, tubes, and, eventually, silica rods. Several monolithic stationary phases are now commercialized and some of their features are summarized in Table I. A very detailed account of these materials can be found in a monograph (3) and several excellent review articles (4–9).


Table I: List of current commercial monolithic polymer-based stationary phases for HPLC separations
Macroporous Monolithic Disks
The disk format is one of the first useful monolithic stationary phases originally designed for the rapid separation of proteins (10). In the mid 1980s, Belenkii and coworkers (11) studied chromatography of proteins in gradient elution mode using stationary phases with a variety of chemistries and column geometries and found that only a certain, often only a very short, distance is required to achieve good separation. This finding resulted in the concept of short separation beds. Because it was very difficult to create such beds from particulate sorbents due to irregularities in packing density and excessive channeling, a new monolithic stationary phase in disk format has been developed by Svec and Tennikova (12–14) at the Institute of Macromolecular Chemistry in Prague, Czech Republic. This then enabled the predicted very fast separations. Typically, the monolithic material is prepared in a flat or tubular mold, the sheet or cylinder removed from the mold, and the porous polymer punched or sliced to obtain up to 3-mm-thin disks. These are then placed in a specifically designed cartridge. In the current commercial implementation called CIM-Disks produced by BIA Separations (Ljubljana, Slovenia), the disk is embedded in a polyolefin ring that forms an impermeable sidewall as shown in Figure 1a and its color indicates specific chemistry of the disk. Although the monoliths are sufficiently mechanically stable to be handled easily, this ring also serves to reinforce the disks and prevent fraying of their edges. An additional benefit is that the flat face of the ring enables the disk to be sealed firmly between the bottom and top face of the cartridge without exercising excessive force on the porous polymer monolith. In contrast to the cartridge, the disks are disposable. The proprietary cartridge is designed to allow insertion of a single disk or several disks. This disk-stacking ability has led to the development of the multidimensional separation process called conjoint liquid chromatography in which disks with different chemistries are used simultaneously. This technique would be difficult to implement with traditional columns. The performance of such a multidisk system was demonstrated with the rapid separation of two proteins and IgG in a single cartridge containing three disks provided with two different ligands, QE (strong anion exchange) and Protein G (affinity), respectively (15).


Figure 1
From the very beginning, monolithic disks exhibited an excellent performance in the rapid separations of proteins and nucleic acids using gradient elution (16). Most of the generic disks are prepared from reactive monomers such as glycidyl methacrylate. The epoxide group is modified subsequently to afford desired interacting functionalities. These modifications enable extension of the separations using disks to a number of modes such as reversed phase, ion exchange, hydrophobic interaction, and bioaffinity chromatography. For example, Peterka modified the CIM disk with iminodiacetic acid, loaded it with Cu(II) ions, and used for the purification of proteins from crude cell extracts (17). An excellent dynamic loading capacity of 18 and 30 mg/mL for proteins LK-801 and GFP-6His as well as a high purity of the separated products demonstrated the advantages of the monolithic disks.
Rigid Macroporous Polymer Monolithic Columns
In the early 1990s, Svec and Fréchet (18) introduced new macroporous polymer monolithic columns formed by a very simple in situ "molding" process. In contrast to disks portrayed previously, these monoliths are polymerized within a tube such as a chromatographic column or capillary in which they remain all the time after the preparation is completed. These monolithic columns are available from Dionex (Sunnyvale, California) under the trade name SwiftPro in numerous formats with functional groups suitable for reversed-phase and ion-exchange chromatography of proteins and polynucleotides.
In addition to the original stainless steel and glass analytical size columns, monolithic columns for high performance liquid chromatography (HPLC) also are now available in capillaries (19–25). This format is readily compatible with mass spectrometry (MS) and therefore is becoming very popular. It is much easier to polymerize liquid precursors and form the stationary phase in situ within a capillary than packing it efficiently with microparticles. Toll and colleagues recently compared separations achieved using a capillary monolithic and packed column for the separation of a complex peptide mixture (26). Figure 2 clearly demonstrates the advantages of the monolith. Monolithic capillary columns made of styrene and divinylbenzene can be purchased from Dionex.


Figure 2
Several methods such as direct copolymerization, chemical modification, and grafting of existing monolith are now available for the preparation of monolithic stationary phases with desired surface chemistry. For example, directly polymerized poly(styrene–co-divinylbenzene) monoliths proved to be an excellent stationary phase for the very fast separation of peptides and proteins (27). Similarly, Eeltink and colleagues prepared alkyl methacrylate-based monolithic columns by direct copolymerization in a capillary and used them for the separation of peptides from protein digests (28). Another option is the preparation of a monolith with reactive functionality and its subsequent modification to afford stationary phases for a variety of separation modes. In this technique, each single reactive site affords one new functionality. Among others, glycidyl methacrylate-based monoliths have been used mainly in these applications (29). Yet another method that enables both preparation of monoliths and modulation of their surface chemistries relies on UV-light triggered processes. Using capillaries with UV transparent PTFE coating, the polymerization can be initiated by irradiation with light and proceeds faster than its thermally-initiated counterpart (30,31). The chemistries of these columns are dictated by the choice of monomers used for their preparation that have to be UV transparent (32). The desired surface chemistry is then obtained via grafting reactions initiated again by UV light (33). One of the major advantages of grafting is that it affords monoliths in which each surface site provides for numerous functionalities, thus, dramatically increasing the column loading capacity.


Figure 3
To observe the effect of time on performance of monolithic capillary columns, we used a single capillary column for the model separation of a mixture of proteins consecutively repeated over 3500 times (34). The chromatograms shown in Figure 3 were recorded during 2500 injections, yet no changes in retention were observed. This study has been extended to demonstrate batch-to-batch and column-to-column repeatability (34). Three batches each including five columns were prepared using both thermally and photoinitiated polymerization. Table II presents the results demonstrating excellent repeatability of the process and column properties.


Table II: Retention times for three model proteins using 30 different monolithic columns prepared via thermally and photochemically initiated polymerization. Conditions: monolithic capillary column 20 cm 3 100 mm I.D.; mobile phase A 2% formic acid in 98:2 water-acetonitrile mixture, mobile phase B 2% formic acid in acetonitrile; gradient from 100% A to 50% B in A in 4 min; flow rate 4 mL/min.
Monolithic Polymer Layer Open Tubular (PLOT) Capillary Columns
Although 100–200 μm i.d. capillary columns offer numerous advantages compared to analytical size columns, they are still large if the amount of sample is very small. Therefore, use of narrow bore capillaries is desirable to reduce band dilution and enhance ionization efficiency in electrospray ionization (ESI)-MS. The preparation of monolith in capillaries with internal diameters less than 20 μm requires specific techniques. Open fused-silica tubes alone, as the other extreme, do not provide for desired retention. This is why two groups recently studied narrow-bore capillary columns with a thin monolithic polymer layer covering the wall of an open tubular column (35–37). The 28-cm-long poly(butyl methacrylate-co -ethylene dimethacrylate) columns were grafted with (2-[methacryloyloxy]ethyl)-trimethylammonium chloride and used for the separations in pressurized capillary electrochromatography (CEC) mode. Despite the very short separation times of less than 90 s, a high column efficiency of almost 200,000 plate/m could be achieved (35). Karger's group used 10-μm i.d. capillaries with porous poly(styrene–co-divinylbenzene) layer for the ultrasensitive proteomic analysis (36,37). The small resistance to flow allowed them to use capillary columns as long as 3.2 m and to achieve an excellent separation of tryptic digest in 50 min with a peak capacity of 400. An even longer column (4.2 m) was used for the separation of a microorganism digest, shown in Figure 4. Over 3000 unique peptides covering 566 distinct Methanosarcina acetivorans proteins were identified from a 50-ng in-gel digest.


Figure 4
Tubular Monolithic Columns with Radial Flow
Large monolithic columns are desirable for the separations on preparative scale. However, free radical polymerization that is used for the preparation of porous polymer monoliths is an exothermic process that creates heat. While this does not appear as a problem in the preparation of capillaries and analytical scale monolithic columns, the accurate control of the polymerization temperature for larger size monoliths is far more problematic (38). The unstirred nature of the polymerization within the confines of a mold leads to a decreased capacity to dissipate the heat of polymerization effectively. In addition to an overall deviation from the desired polymerization temperature, the temperature also can vary in magnitude across the contents of the mold in the radial direction. In light of the demonstrated effect of the polymerization temperature on the porosity of the resulting polymer, any significant variation in temperature within the mold leads to monoliths with heterogeneity in their pore structures and significantly reduced performance.
This problem was alleviated elegantly by the preparation of monoliths in an annular shape shown in Figure 1b (39). Because the walls of the annulus are thinner than those of a solid cylinder with an equal outer diameter, control of the temperature is facilitated. This approach also enables independent variation of both thickness of the tube wall and its inner diameter. Thus, while keeping the thickness of the wall constant, a significant increase in volume can be achieved easily. Another advantage of this approach includes the preparation of series of tubes that fit each in the other. This sort of "telescopic" format enables construction of rather large separation devices that would be very difficult to obtain in a single polymerization step. For example, tubular columns called CIM-Tubes (BIA Separations) with a volume of up to 8 L operating in the radial direction at a flow rate of up to 10 L/min have been commercialized by BIA and used for the rapid preparative separations of 200 g of proteins in a single run. These tubes also were used in a good manufacturing practice (GMP) certified industrial purification of plasmid DNA for therapeutic applications. The largest unit enables recovery of 10–50 mg of a pure pDNA product in a single run.
Monolith for On-Line Sample Preparation
Analytical processes including complex matrices such as biological fluids often require a sample preparation step before the separation and detection. While this step frequently is carried out off-line, on-line solid-phase extraction (SPE) combined with HPLC–MS presents numerous advantages (40). It enables faster and fully automated analyses, reduces human contact with contaminated or hazardous samples, and potentially increases sensitivity because the complete sample can be injected. Direct injection of biological fluids such as serum or plasma in columns packed with conventional chromatographic supports often leads to rapid deterioration of the performance. In contrast, monolithic supports are suitable for preconcentration of compounds from biological fluids. The first paper describing polymer-based monoliths applied to SPE was published by our group in 1998 (41) and was followed by numerous other researchers thereafter (42). The ability to prepare the monolith in a specific location via the photoinitiated polymerization, facilitates their broad applications as in-column preconcentration units in CEC, capillary electrophoresis (CE), and nano-HPLC. For example, Wei and colleagues (43) used poly(methacrylic acid-co -ethylene dimethacrylate) monolith for microextraction, and a field-enhanced sample injection preconcentration technique was proposed for sensitive capillary electrophoresis of ephedrine and pseudoephedrine in human plasma and urine. A hydrolyzed poly(glycidyl methacrylate-co -ethylene dimethacrylate) monolith in capillary was used for solid-phase microextraction of polar compounds such as nitrophenols (44).


Table III: Comparison of digestion of bovine serum albumin (BSA) using trypsin in solution and immobilized on monolithic support
Enzyme Immobilization Using Monolithic Supports
Present proteomics uses two major approaches: top-down techniques first involving the separation of proteins followed by digestions of specific fractions using a proteolytic enzyme and mass spectrometric analysis, and bottom-up methods characterized by the initial digestion of all proteins in the sample, followed by separation of the peptide fragments and their mass spectrometric analysis. A subsequent comparison of the mass distribution profile within an appropriate database is then used to identify the original proteins. No matter what approach is chosen, proteolytic digestion of the protein to easier-to-define peptides is always included in the protocol. Monolithic supports were found extremely suitable for immobilization of proteolytic enzymes and preparation of highly active enzymatic reactors (45–48). The major advantage characteristic of the monolithic support is the outstanding mass transfer it enables due to the convective flow through the pores. This is well documented with the significantly enhanced rates of tryptic digestion of proteins compared to the reaction in solution. Table III compares our results for digestion of bovine serum albumin with both soluble and immobilized trypsin. While digestion with the former requires 24 h at 37 °C, the latter enables a similar degree of digestion to be achieved in less than 5 min even at a lower temperature of 22 °C. The typical sample size in proteomics is very small. Therefore, miniature immobilized enzyme reactors placed in capillaries or microfluidic devices are highly suitable for proteomic studies. An additional benefit is the option to link the microreactor directly to mass spectrometer. Figure 5 shows our matrix-assisted laser desorption ionization (MALDI) MS spectrum of BSA digest prepared using trypsin immobilized on monolith with grafted poly(4,4-dimethyl-1-vinylazlactone) chains.

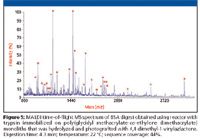
Figure 5
Monoliths in Microfluidics
In contrast to columns, the microfabricated devices feature a network of microchannels etched in glass or imprinted in a polymer plate that are designed to enable much smaller sample volumes to be analyzed at an increased speed and permitting a large number of analyses to be performed simultaneously, thus, increasing the overall throughput. Because the surface-to-volume ratio typical of open channel does not support high-performance separations, filling the channel with the monolithic materials enables the user to place a suitable stationary phase in the channel. In contrast to the tedious packing of channels with particles, monoliths are prepared from liquid precursors that facilitate filling the channel. The monolith then can be placed in an exactly-defined part of the chip using polymerization photoinitiated through a mask. Although most of the reports concern separations in the reversed-phase mode, affinity chromatography and electrochromatography also have been demonstrated (49).
Conclusion
Despite the ever-growing number of publications, monolithic columns still remain Cinderella among current stationary phases for HPLC. However, recent achievements presented previously indicate that there is a lot of space available for the expansion of this entirely new class of materials. The large amount of experimental work increasing every day, along with the commercial availability of monolithic columns, confirms the great potential of monoliths (50,51). The unique properties of monolithic column, in particular, the ease of their preparation, the tolerance to high flow rates, and the unprecedented speed of the chromatographic separations that can be achieved at acceptable back pressures, make them in some applications superior to the more common columns packed with beads.
When the range covered by the monolithic technologies will be extended and monoliths will successfully compete with the well-established particulate column packings is just a question of time. What is currently preventing the monolithic columns from even wider acceptance? Certainly not poor reproducibility often mentioned by their critics. Our results shown previously confirmed that this claim is false. First of all, they compete with the many decades old packed columns. The major "selling" feature, the high-speed separations, might not be attractive to those who inject two samples a day. In contrast, laboratories such as those in pharmaceutical or biotechnology industry, where high-throughput is indispensable, certainly can appreciate the speed enabled by monoliths not only in separations but also in digestion. However, a significant problem arises from the limited number of companies producing monolithic columns. Industrial laboratories do not like sole sources; in particular, not for validated processes. And last but not least, the current choice of sizes and chemistries of monolithic columns does not match that of the "classical" packed counterparts. Some formats such as longer 1–2 mm i.d. columns are not available commercially and the only really large-scale monolithic column is the 8-L tubular unit with axial flow.
This article describes a small number of applications of porous polymer monoliths. However, a large promise of these flow-through materials also can be seen in other areas such as electrochromatography, gas chromatography, heterogeneous catalysis, and combinatorial chemistry (3).
Acknowledgment
The work carried out in the Molecular Foundry (http://foundry.lbl.gov/) was supported by the Director, Office of Science, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
References
(1) J.J. Meyers and A.I. Liapis, J. Chromatogr., A 852, 3–23 (1999).
(2) F.C. Leinweber, D. Lubda, K. Cabrera, and U. Tallarek, Anal. Chem. 74, 2470–2477 (2002).
(3) F. Svec, T. B. Tennikova, and Z. Deyl, Eds., Monolithic Materials: Preparation, Properties, and Applications, (Elsevier, Amsterdam, 2003).
(4) F. Svec, J. Sep. Sci. 27, 747–766 (2004).
(5) K. Miyabe and G. Guiochon, J. Sep. Sci. 27, 853–873 (2004).
(6) K. Mistry and N. Grinberg, J. Liquid Chromatogr. Rel. Tech. 28, 1055–1074 (2005).
(7) G. Guiochon, J. Chromatogr., A 1168, 101–168 (2007).
(8) M.R. Buchmeiser, Polymer 48, 2187–2198 (2007).
(9) K.W. Ro, R. Nayak, and D.R. Knapp, Electrophoresis 27, 3547–3558 (2006).
(10) D. Josic and A. Strancar, Ind. Eng. Chem. Res. 38, 333–342 (1999).
(11) B.G. Belenkii, A.M. Podkladenko, O.I. Kurenbin, V.G. Mal''''tsev, D.G. Nasledov, and S.A. Trushin, J. Chromatogr. 645, 1–15 (1993).
(12) T.B. Tennikova, F. Svec, and B.G. Belenkii, J. Liq. Chromatogr. 13, 63–70 (1990).
(13) T.B. Tennikova, M. Bleha, F. Svec, T.V. Almazova, and B.G. Belenkii, J. Chromatogr. 555, 97–107 (1991).
(14) F. Svec and T.B. Tennikova, J. Chromatogr. 646, 279–288 (1993).
(15) K. Branovic, G. Lattner, M. Barut, A. Strancar, D. Josic, and A. Buchacher, J. Immunol. Meth. 271, 47–58 (2002).
(16) D. Josic, J. Reusch, K. Loster, O. Baum, and W. Reutter, J. Chromatogr. 590, 59–76 (1992).
(17) M. Peterka, M. Jarc, M. Banjac, V. Frankovic, K. Bencina, M. Merhar, V. Gaberc-Porekar, V. Menart, A. Strancar, and A. Podgornik, J. Chromatogr., A 1109, 80–85 (2006).
(18) F. Svec and J.M.J. Frechet, Anal. Chem. 54, 820–822 (1992).
(19) R.E. Moore, L. Licklider, D. Schumann, and T.D. Lee, Anal. Chem. 70, 4879–4884 (1998).
(20) X. Huang, S. Zhang, G.A. Schultz, and J.D. Henion, Anal. Chem. 74, 2336–2344 (2002).
(21) P. Holdsvendova, P. Coufal, J. Suchankova, E. Tesarova, and Z. Bosakova, J. Sep. Sci. 26, 1623–1628 (2003).
(22) D. Moravcova, P. Jandera, J. Urban, and J. Planeta, J. Sep. Sci. 26, 1005–1016 (2003).
(23) H. Oberacher and C.G. Huber, Trends Anal Chem. 21, 166–174 (2002).
(24) A.R. Ivanov, L. Zang, and B.L. Karger, Anal. Chem. 75, 5306–5316 (2003).
(25) S. Eeltink, W.M.C. Decrop, G.P. Rozing, P.J. Schoenmakers, and W.T. Kok, J. Sep. Sci. 27, 1431–1440 (2004).
(26) H. Toll, R. Wintringer, U. Schweiger-Hufnagel, and C.G. Huber, J. Sep. Sci. 28, 1666–1674 (2005).
(27) S. Xie, R.W. Allington, F. Svec, and J.M.J. Fréchet, J. Chromatogr., A 865, 169-174 (1999).
(28) B. Eeltink, L. Geiser, F. Svec, and J.M.J. Fréchet, J. Sep. Sci. 30, 2814–2820 (2007).
(29) S. Xie, R.W. Allington, J.M.J. Fréchet, and F. Svec, Adv. Biochem. Eng. Biotechnol. 76, 88–123 (2002).
(30) C. Viklund, E. Ponten, B. Glad, K. Irgum, P. Horsted, and F. Svec, Chem. Mater. 9, 463–471 (1997).
(31) C. Yu, F. Svec, and J.M.J. Fréchet, Electrophoresis 21, 120-127 (2000).
(32) D. Lee, F. Svec, and J.M.J. Fréchet, J. Chromatogr., A 1051, 53–60 (2004).
(33) T. Rohr, E.F. Hilder, J.J. Donovan, F. Svec, and J.M.J. Fréchet, Macromolecules, 36, 1677–1684 (2003).
(34) L. Geiser, S. Eeltink, F. Svec, and J.M.J. Fréchet, J. Chromatogr., A 1140, 140–146 (2007).
(35) S. Eeltink, F. Svec, and J.M.J. Frechet, Electrophoresis 27, 4249–4256 (2006).
(36) G. Yue, Q. Luo, J. Zhang, S.L. Wu, and B.L. Karger, Anal. Chem. 79, 938–946 (2007).
(37) Q. Luo, G. Yue, G.A. Valaskovic, Y. Gu, S.L. Wu, and B.L. Karger, Anal. Chem. 79, 6174–6181 (2007).
(38) E.C. Peters, F. Svec, and J.M.J. Fréchet, Chem. Mater. 9, 1898–1902 (1997).
(39) A. Podgornik, M. Barut, A. Strancar, D. Josic, and T. Koloini, Anal. Chem. 72, 5693–5699 (2000).
(40) J.L. Veuthey, S. Souverain, and S. Rudaz, Ther. Drug Monit. 26, 161–166 (2004).
(41) S. Xie, F. Svec, and J.M.J. Fréchet, Chem. Mater. 10, 4072–4078 (1998).
(42) F. Svec, J. Chromatogr., B 841, 52–64 (2006).
(43) F. Wei, M. Zhang, and Y.Q. Feng, J. Chromatogr., B 850, 38–44 (2007).
(44) Y. Wen and Y.Q. Feng, J. Chromatogr., A 1160, 90–98 (2007).
(45) D. Josic and A. Buchacher, J. Biochem. Biophys. Meth. 49, 153–174 (2001).
(46) J. Krenkova and F. Foret, Electrophoresis, 25, 3550–3563 (2004).
(47) G. Massolini and E. Calleri, J. Sep. Sci. 28, 7–21 (2005).
(48) F. Svec, Electrophoresis, 27, 947–961 (2006).
(49) F. Svec, T. B. Stachowiak, in Handbook of Capillary and Microchip Electrophoresis and Associated Microtechniques , J. Landers, Ed. (Taylor & Francis, Boca Raton, 2007), pp. 1297–1326).
(50) S. Miller, Anal. Chem. 76, 99A–101A (2004).
(51) F. Svec and C.G. Huber, Anal. Chem. 78, 2100–2107 (2006).




















New TRC Facility Accelerates Innovation and Delivery
April 25th 2025We’ve expanded our capabilities with a state-of-the-art, 200,000 sq ft TRC facility in Toronto, completed in 2024 and staffed by over 100 PhD- and MSc-level scientists. This investment enables the development of more innovative compounds, a broader catalogue and custom offering, and streamlined operations for faster delivery. • Our extensive range of over 100,000 high-quality research chemicals—including APIs, metabolites, and impurities in both native and stable isotope-labelled forms—provides essential tools for uncovering molecular disease mechanisms and exploring new opportunities for therapeutic intervention.
New Guide: Characterising Impurity Standards – What Defines “Good Enough?”
April 25th 2025Impurity reference standards (IRSs) are essential for accurately identifying and quantifying impurities in pharmaceutical development and manufacturing. Yet, with limited regulatory guidance on how much characterisation is truly required for different applications, selecting the right standard can be challenging. To help, LGC has developed a new interactive multimedia guide, packed with expert insights to support your decision-making and give you greater confidence when choosing the right IRS for your specific needs.

