Enhanced Stability Stationary Phases for HPLC
Special Issues
This article presents an overview of high performance liquid chromatography stationary phases with enhanced stability at high pH, focusing on the methods by which they were prepared. Among the many alternatives, the authors introduce reversed phases based upon metallized silica supports that show superior performance during stability testing at high pH, when compared with conventional C18 phases based upon bare silica.
This article presents an overview of high performance liquid chromatography stationary phases with enhanced stability at high pH, focusing on the methods by which they were prepared. Among the many alternatives, the authors introduce reversed phases based upon metallized silica supports that show superior performance during stability testing at high pH, when compared with conventional C18 phases based upon bare silica.
Since the first report of chemically bonded silica-based phases for high performance liquid chromatography (HPLC), almost 40 years ago (1), remarkable improvements in the stability of HPLC stationary phases have been achieved, as described by some extensive reviews published in recent years on the chemical and thermal stability of these phases (2–4). In this article, we will focus on some of the advances that were successful in enhancing the chemical stability of phases at high pH and at elevated temperatures, including some that have been described recently.
Traditional Modifications of Silica Using Silanes
The chemical modification that is performed conventionally under anhydrous conditions using monofunctional silanes can be replaced by reactions in the presence of well-controlled amounts of water using trifunctional C18 silanes (5). The resulting polymeric C18 phases provide better stability at high pH, compared to monomeric phases. However, the surface coverage as well the amount of residual silanols is less reproducible, compared to monomeric silica phases. Another disadvantage with the polymeric C18 phases is that densely bonded polymer chains on the surface lead to slower mass transfer kinetics and the optimal flow rates are generally lower. Polymeric C18 phases can be purchased from several different column manufacturers and they are considered very useful for separations that require higher shape selectivity such as the separation of polynuclear aromatic hydrocarbons.



In 1989, silanization using bidentate silylant agents was proposed by Kirkland and coworkers (6). The bidentate reagents have two silicons bridged by –CH2CH2– or –CH2CH2CH2– groups and also have long n-alkyl pendant groups (7). These modified phases possess extended high pH and temperature tolerances because the bidentate silane species better protect the silica support. Column stability is measurably better when compared with C18 phases based upon monofunctional silane chemistry. This phase, commercialized by Agilent Technologies (Santa Clara, California), is appropriate for separation of basic compounds using high pH mobile phases and at temperatures up to 40 °C at high pH.
Stationary Phases Using Alternative Supports
Possible candidates to substitute for silica as a support for problematic separations are porous organic matrices like graphitic carbon, some polymers and other porous inorganic materials, such as ZrO2, TiO2, Al2O3 that, in principle, should have the same chromatographic properties as silica. Another group includes organic-inorganic hybrid supports whose structure still resembles that of inorganic silica but, at the same time, combines some features of polymeric materials. Similarly, metallized silica supports are also a promising alternative.
Reversed Phases Based Upon Metal Oxides
Reversed phases based upon zirconia have been commercialized by ZirChrom (Anoka, Minnesota) since the 1990s. ZrO2 exhibits excellent high pH stability in comparison with silica (8). One limitation in the early stage of this technology was the difficulty in synthesizing ZrO2, TiO2 and Al2O3 particles with appropriate particle size and shape and having surface areas adequate for use in HPLC. Another problem is organofunctionalization. The first experiments to modify these oxides by using conventional silane chemistries were not promising, due to lower coverages, mainly because of the lower number of hydroxyl groups on these oxides when compared to silica surfaces, and to the rapid loss of the bonded phase (9,10) as their surface chemistry is significantly different from silica. For example, the great majority of the zirconyl groups on porous zirconia are in a bridged form on the surface and, as a consequence, these groups are less reactive toward silanization when compared to free silanols on the silica surface (11). However, the use of polymeric (for example, polybutadiene, polystyrene) and carbon coatings proved to be a successful approach to overcome these problems for the zirconia-based phases (12). Recent reports indicate that zirconia phases are well suited for use in high temperature HPLC, even up to 200 °C, using water as the mobile phase (13,14).
Silanization using the trifunctional silylant (CH3O)3 Si-H followed by hydrosilylation with alkenes was explored for the preparation of phases based upon alumina (15), and then extended to titania (16,17). Chemically bonded reversed phases based upon alumina also have been prepared by other reactions with organosilanes (18), but, similar to the phases based upon zirconia, the commercial phases are prepared by coating the surface with a hydrophobic polymer such as polybutadiene and, less commonly, with polyalkylsiloxanes (19). Other polymer coatings also have been investigated using a prior modification of the alumina surface with maleic acid before adding a long alkyl chain. (20). The retention and selectivity of alumina phases has been compared with other stationary phases recently (21). Up to the present, there is a lack of information about the stability of stationary phases based upon titania (22), possibly because there is only one reversed phase, a polyethylene-coated TiO2, which is commercialized by ZirChrom. Polybutadiene-coated alumina phases are commercially available from Merck KGaA (Darmstadt, Germany).
Reversed Phases Based Upon Graphitic Carbon
Porous graphitic carbon, initially suggested more than 20 years ago, also has some attractive properties as a support because it is not affected by physical or chemical degradation at high temperature regardless of the composition of the mobile phase (23). Phases based upon bare carbon are convenient for high temperature HPLC, being a promising alternative to speed up separations (24). On the other hand, direct modification of the surface is almost impossible and the resulting nonpolar phases show less than ideal stability. Carbon-based phases have been commercialized by Thermo Fisher Scientific (Waltham, Massachusetts) and many applications have been reported, including the separation of lipids (25), triacylglycerols (26), organic explosives (27), and polar phenolic compounds (28). More recently, surfactant-coated graphitic carbon-based stationary phases have been used in anion-exchange chromatography (29).
Reversed Phases Based Upon Polymeric Supports
Contrary to silica, the chemical stability of polymer-based phases is not affected by the pH or by the presence of buffers in the mobile phase. However, these phases have significantly lower efficiencies for small molecules as well as lower pressure and temperature resistances, as well as swelling and shrinking problems with certain organic solvents. Modern polymeric stationary phases consist of highly crosslinked porous copolymers that can be prepared with a broad range of functionalities in a single step by the copolymerization of functional monomers. Although simple, this approach requires significant optimization of the preparation process to control the porosity of the beads each time a new monomer is used. Polymer phases are commercialized by various column manufactures and their most common application is for the separation of pressure and temperature sensitive biological molecules. They also are used often for ion-exchange and size-exclusion separations. Polymer phases are commercialized by several different suppliers, including Bio-Rad (Hercules, California), Dionex (Sunnyvale, California), Hamilton (Reno, Nevada), Phenomenex (Torrance, California), and Sigma/Aldrich Supelco (Bellefonte, Pennsylvania). Synthetic polymers are also very promising as stationary phases in monolithic capillary columns for the emerging separation techniques of micro HPLC and capillary electrochromatography (CEC) and also for applications on microchips (30) because the phases can be generated in situ.
Reversed Phases Based Upon Hybrid Silica
The concept of hybrid silica for HPLC was introduced in the seventies by Unger and Scharf (31) but was launched commercially only in 2000 by Waters (Miliford, Massachusetts). These phases present the same efficiency and pressure resistance as phases based upon pure silica, combined with significantly wider pH stabilities (32). The breakthrough in this technology was the production of spherical mesoporous organic–inorganic silica particles, synthesized by the sol-gel process from a mixture of tetraethoxysilane and methyltriethoxysilane in an appropriate solvent containing a porogenic agent. As a consequence, some of the hydroxyls are replaced by a methyl group and the final silanol content is lower when compared to sol-gel silicas prepared from pure tetraethoxysilane. After modification with C18, the residual silanol concentration is even lower, providing excellent peak shapes for highly basic compounds. Another advantage of the limited number of residual silanols on the hybrid silica surface is improved hydrolytic stability at high pH, because it is the unreacted silanols on the pure silica surface that are the points at which dissolution starts. On the other hand, the C18 coverage of these hybrid phases is lower than that of monofunctional C18 silica phases.
Four years later, the production of hybrid particles incorporating a bidentate silane was reported (33). By replacing methyltriethoxysilane with bis(triethoxysilyl)ethane, where the –CH2CH2- fragment is attached to two triethoxysilyl groups, the resulting hybrid surface has bridging groups. These phases are considered the second generation of hybrid phases and exhibit even better mechanical (34), thermal, and hydrolytic stability (35) than the hybrid phases prepared with methyl substitution. To date, this second generation of hybrid phases is offered only by Waters. A recent study on the stability of this phase using 1.7-μm particles under ultrahigh-pressure liquid chromatography (UHPLC) conditions (90 °C and a pressure drop of 600 bar) for the separation of parabens in a nonbuffered mobile phase reported that the phase was stable up to 500 injections (equivalent to 7500 column volumes) (36).
Reversed Phases Based Upon Metallized Silicas
Silicas metallized with titanium or zirconium oxide have been explored in our research group as supports for the preparation of stationary phases having polysiloxanes and other preformed polymers immobilized on the surface (37–42), using a strategy first explored in our laboratory using a bare silica surface (43). More recently, metallized supports have proven to be very useful to prepare chemically bonded C18 phases (44–49).
The goal of this technology is to make the support less sensitive to dissolution at high pH due to the presence of more hydrolytically stable layers of a metal oxide. These layers can be grafted on the surface using metal alkoxides, preserving the particle morphology and the mesoporous structure of the bare silica. Although the chemical properties of the metallized support are different, modifications using conventional silane chemistry provide stationary phases with structures similar to the one shown in Figure 1.

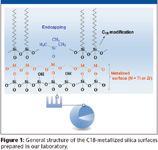
Figure 1
Evidence for the success of using a thin layer of another oxide between silica and alkylsilyl ligands was first pointed out by Stout and DeStefano in 1985 (50) They reported a diol phase based upon zirconized silica prepared by reaction with ZrOCl2 before silanization. Interestingly, even after thirty years, this zirconium-stabilized-silica diol phase is still commercialized by Agilent Technologies for the separation of biological macromolecules.
A decade later, the idea of having stationary phases based upon mixed oxide silicas also was explored by Kineko and coworkers, in a study in which composites of silica with alumina, titania, zirconia, or magnesia were prepared by a coprecipitation method then used for normal phase chromatography (51).
In our group, the investigation of metallized silicas as supports for chemically bonded C18 phases began in 2003, using titanized silica. The first attempts to modify the surface using octadecyltrichlorosilane were not successful, but using a trimethoxy silane, the C18 coverages of the titanized supports were similar to those obtained for unmodified silica (47). However, due to the presence of the titanium oxide layer, the acidities of the residual hydroxyl groups were increased.
To illustrate the applicability of the SiTiC18 phase for real world samples, Figure 2 shows the separation of two highly basic pharmaceuticals at pH 12. These drugs are usually difficult to separate using conventional C18 phases based on silica because, at neutral pH, the peaks exhibit considerable tailing while, at low pH, they are protonated and are poorly retained. With high pH mobile phases, they are separated as free base species, but the bare silica support starts to dissolve, rapidly provoking column failure. However, the titanized-silica reversed phase has an extended column lifetime under these basic conditions.

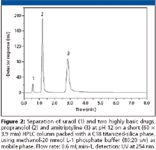
Figure 2
A further step was the preparation of titanized phases with urea polar-embedded groups. The titanized surface was modified using a urea-trialkoxysilane, also synthesized in our laboratory, to have the urea groups near to the surface, as shown in Figure 3, to minimize the undesirable interactions of the acidic residual groups with bases. The retention factors for weakly basic compounds were lower with the polar-embedded phase, however, some tailing still was observed when using nonbuffered mobile phases (46). This phase also was used successfully for the separation of propranolol and amitriptyline at pH 11, using a buffered mobile phase (Figure 4).

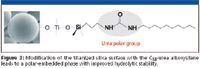
Figure 3
The influence of the TiO2 content on performance and stability at high pH of some C18 titanized phases also was studied (47). A C18 doubly titanized stationary phase was prepared by performing two titanization reactions before the silanization and endcapping steps. The higher TiO2 content on the surface did not affect the overall performance in terms of mass transfer because the efficiencies for neutral compounds and the optimal flow rates were similar to those of the singly titanized phase.

Figure 4
Results of recent studies with octadecyl phases bonded to zirconized silica also have been reported (49). Similar to the titanized phases, the ZrO2 modified silicas were prepared by reaction of the bare silica with a zirconium alkoxide, followed by C18 silanization and endcapping. The zirconized phases were evaluated using the Tanaka and the Engelhardt test protocols. As observed for our titanized silica phases and for the commercial phases based upon zirconia, the zirconized C18 phase also exhibited higher retention for basic analytes in non-buffered mobile phases. For practical use, the separation of bases on the zirconized-silica phase requires the addition of buffers or amines to the mobile phase to minimize the acid–base interactions during the separation.
SiO2-ZrO2 as well as SiO2-TiO2 materials also have been investigated by Chen and coworkers (52–54). In their work, the metal oxide nanoparticles are attached using a layer-by-layer approach that involves the adsorption of a surfactant onto silica, followed by deposition of a zirconia or titania sol. This process is repeated several times and the surfactant eventually is burnt off. The support is then modified with octadecyltrichlorosilane. Nonetheless, their chromatographic efficiencies were quite low, when compared with the results obtained with our metallized silica phases.
Extension of the concept of metallized phases to monolithic silica technology is almost unexplored. One example describes the deposition of TiO2 onto monolithic silica for the HPLC separation of phosphorylated analytes (55). The efficiencies were slightly lower, but this could be improved by an optimization of the titanization procedure. Zirconia-coated silica monoliths also were modified with alkylphosphonates and were used for the separation of some neutral compounds by CEC. Stability experiments were very limited but, interestingly, the electroosmotic flow differed significantly from that of an unmodified silica monolith (56).
Stability Evaluations of Some New Reversed Phases
Stability testing is very important in the development of novel stationary phases. However, it is difficult to compare the data presented by different column manufacturers or between different research groups because the conditions often vary considerably. For high pH stability tests, the use of triethylamine or pyrrolidine to increase the pH is not uncommon. Nonetheless, these amines are known to be less aggressive to the chromatographic support than phosphate and carbonate salts (57). Another important point is that some column manufacturers report only the retention factor data for tests at high pH. However, because these values do not change significantly, while column collapse at high pH is associated directly with silica dissolution and the consequent formation of voids in the column. These attributes are characterized by abrupt decreases in plate numbers as well as steep increases in tailing factors before changes are seen in the retention factors.
To compare the column life of the different stationary phases prepared by silanization of the metallized silicas prepared in our laboratory, we adopted a standardized stability test using a temperature of 50 °C and a nominally pH 10 mobile phase containing phosphate. Both conditions are known to increase the dissolution of a silica support. As shown in several publications (40,44,45,49), this stability test can provide repetitive results and many of the stationary phases developed in our laboratory have been investigated and compared to some commercial phases. To summarize some of our more significant findings, Figure 5 illustrates the retention factors and the efficiencies measured using acenaphthene as probe during stability testing for the titanized and zirconized C18 phases (SiTiC18 and SiZrC18, respectively) and for two commercial phases, a monomeric C18 phase based upon silica and a hybrid C18 column.


Figure 5
The zirconized silica phase was more stable than either the commercial C18 phase or the titanized silica phase. The hybrid C18 column gave the best stability results. Similar trends also have been observed for the metallized phases prepared by thermal immobilization of several polysiloxanes onto metallized silicas (37).
Conclusions
Chemically bonded C18 phases based upon metallized silicas are an alternative to the many technologies that have been proposed to enhance the durability of HPLC columns. Improvements in the stability of the metallized phases are still a subject of investigation in our research group, exploring alternative routes for the metalization of silica as well as the use of other silylant agents to bond the alkyl chains to the surface, with the goal of obtaining excellent pH stability in stationary phases for reversed-phase HPLC.
César R. Silva and Carol H. Collins, Instituto de Química, Universidade Estadual de Campinas, Campinas, São Paulo, Brazil. Please direct correspondence to chc@iqm.unicamp.br
References
(1) R.E. Majors, LCGC 12, 508–512 (1994).
(2) H.A. Claessens and M.A. van Straten, J. Chromatogr., A 1060, 23–41 (2004).
(3) J. Nawrocki, C. Dunlap, A. McCormick, and P.W. Carr, J. Chromatogr., A 1028, 1–30 (2004).
(4) H. Claessens, TrAC, Trends Anal. Chem. 20, 563–583 (2001).
(5) R.W. Fairbank and M.J. Wirth, J. Chromatogr., A 830, 285–291 (1999).
(6) J.J. Kirkland, J.L. Glajch, and R.D. Farlee, Anal. Chem. 61, 2–11 (1989).
(7) J.J. Kirkland, J.B. Adams Jr., M.A. van Straten, and H.A. Claessens, Anal. Chem. 70, 1998, 4344-4352.
(8) M.P. Rigney, T.P. Weber, abd P.W. Carr, J. Chromatogr. 484, 273–291 (1989).
(9) U. Tründinger, G. Müller, and K.K. Unger, J. Chromatogr. 535, 111–125 (1990).
(10) J. Yu and Z. El Rassi, J. Chromatogr. 631, 91–106 (1993).
(11) C.P. Jaroniec, M. Jaroniec, and M. Krurk, J. Chromatogr., A 797, 93–102 (1998).
(12) J. Nawrocki, C. Dunlap, J. Li, J. Zhao, C.V. McNeff, A. McCormick, and P.W. Carr, J. Chromatogr., A 1028, 31–62 (2004).
(13) R. Smith, J. Chromatogr., A 1184, 441–455 (2008).
(14) T. Teutenberg, J. Tuerk, M. Holzhauser, and S. Giegold, J. Sep. Sci. 30, 1101–1114 (2007).
(15) J.J Pesek, J.E. Sandoval, and M. Su, J. Chromatogr. 630, 95–103 (1993).
(16) J.J Pesek, M.T. Matyska, and J. Ramakrisham, Chromatographia 44, 538–544 (1997).
(17) A. Ellwanger, M.T. Matyska, K. Albert, and J.J. Pesek, Chromatographia 49, 424–430 (1999).
(18) J.J. Pesek and H.D. Lin, Chromatographia 28, 565–568 (1989).
(19) R.V. Arenast and J.P. Foley, Analyst 119, 1303–1314 (1994).
(20) Y. Mao and B.M. Fung, J. Chromatogr., A 790, 9–15 (1997).
(21) P. Jandera, K. Novotná, M.S. Beldean-Galea, and K. Jísa, J. Sep. Sci. 29, 856–871 (2006).
(22) V. Zizkovsk?, R. Kucera, J. Klimes, and J. Dohnal, J. Chromatogr., A, 2008, in press.
(23) L. Pereira, S. Aspey, and H. Ritchie, J. Sep. Sci. 30, 1115–1127 (2007).
(24) R.E. Majors, LCGC 26(1), 16–19 (2008).
(25) A. Hazotte, D. Libong, and P. Chaminade, J. Chromatogr., A 1140, 131–139 (2007).
(26) B. Merelli, M. De Person, P. Favetta, and M. Lafosse, J. Chromatogr., A 1157, 462–466 (2007).
(27) E. Holmgren, H. Carlsson, P. Goede, and C. Crescenzi, J. Chromatogr., A 1099, 127–135 (2005).
(28) J. Vial, M. Hennion, A. Fernandez-Alba, and A. Agüera, J. Chromatogr., A 937, 21–29 (2001).
(29) S.D. Chambers and C.A. Lucy, J. Chromatogr., A 1176, 178–184 (2007).
(30) S. Eeltink and F. Svec, Electrophoresis 28, 137–147 (2007).
(31) K. Unger and B. Scharf, J. Colloid Interface Sci.55, 377–380 (1976).
(32) Y.F. Cheng, T.H. Walter, Z. Lu, P. Iraneta, B.A. Alden, C. Genderau, U.D. Neue, J.M. Grassi, J.L. Carmody, J.E. O'Gara, and R.P. Fisk, LCGC 18, 1162–1170 (2000).
(33) K.D. Wyndhan, J.E. O'Gara, T.H. Walter, K.H. Glose, N.L. Lawrence, B.A. Alden, G.S. Izzo, C.J. Hudalla, and P.C. Iraneta, Anal. Chem. 75, 6781–6788 (2004).
(34) K.D. Patel, A.D. Jerkovich, J.C. Link, and J.W. Jorgenson, Anal. Chem. 76, 5777 (2004).
(35) Y. Liu, N. Grinberg, K.C. Thompson, R.M. Wenslow, U.D. Neue, D. Morrison, T.H. Walter, J.E. O'Gara, and K.D. Wyndham, Anal. Chim. Acta 554, 144–151 (2005).
(36) D.T. Nguyena, D. Guillarme, S. Heinisch, M. Barrioulet, J. Rocca, S. Rudaz, and J. Veuthey, J. Chromatogr., A 1167, 76–84 (2007).
(37) A.M. Faria, I.C.S.F. Jardim, K.E. Collins, and C.H. Collins, J. Sep. Sci 29, 782–789 (2006).
(38) A.M. Faria, K.E. Collins, and C.H. Collins, J. Chromatogr., A 1122, 144–122 (2006).
(39) A.M. Faria, E. Tonhi, K.E. Collins, and C.H. Collins, J. Sep. Sci 30, 1844–1851 (2007).
(40) D.A. Fonseca, K.E. Collins, and C.H. Collins, J. Chromatogr. A 1030, 49–54 (2001).
(41) R.B. Silva, Y. Gushikem, and C.H. Collins, J. Sep. Sci. 24, 129–135 (2000).
(42) L.F.C. Melo, K.E. Collins, C.H. Collins, and I.C.S.F. Jardim, J. Chromatogr., A 869, 49–54 (2001).
(43) T.A. Anazawa and I.C.S.F. Jardim, J. Liq. Chromatogr. 17, 1265–1279 (1994).
(44) C.R. Silva, C. Airoldi, K.E. Collins, and C.H. Collins, LCGC 22, 632–641 (2004).
(45) C.R. Silva, C. Airoldi, K.E. Collins, and C.H. Collins, J. Chromatogr., A 1073, 155–162 (2005).
(46) C.R. Silva, C. Airoldi, K.E. Collins, and C.H. Collins, J. Chromatogr., A 1087, 29–37 (2005).
(47) C.R. Silva, C. Airoldi, K.E. Collins, and C.H. Collins, J. Chromatogr., A 1114, 45–52 (2005).
(48) C.R. Silva, C. Airoldi, K.E. Collins, and C.H. Collins, J. Sep. Sci. 29, 790–800 (2006).
(49) C.R. Silva, C. Airoldi, K.E. Collins, and C.H. Collins, J. Chromatogr., A, 2008, in press.
(50) R.W Stout and J.J. DeStefano, J. Chromatogr. 326, 63–78 (1985).
(51) S. Kaneko, T. Mitsuzawa, S. Ohmori, M. Nakamura, K. Nobuhara, and M. Masatani, J. Chromatogr., A 669, 1–7 (1994).
(52) H. Dun, W. Zhang, Y. Wei, S. Xiuqing, Y. Li, and L. Chen, Anal. Chem. 76, 5016–5023 (2004).
(53) J. Ge, X. Shi, Y. Li, and L. Chen, Chromatographia 63, 25–30 (2006).
(54) J. Ge, Y. Li, and L. Chen, J. Liq. Chromatogr. Rel. Technol. 29, 2329–2339 (2006).
(55) S. Miayazaki, M.Y. Miah, K. Morisato, Y. Shintani, T. Kuroha, and K. Nakanishi, J. Sep. Sci. 28, 39–44 (2005).
(56) Z.G. Shi, Y.Q. Feng, L. Xu, M. Zhang, and S.L. Da, Talanta 63, 593–598 (2004).
(57) J.J. Kirkland, J.W. Henderson, J.J. DeStefano, M.A. van.Straten, and H.A. Claessens, J. Chromatogr., A 762, 97–112 (1997).




















New TRC Facility Accelerates Innovation and Delivery
April 25th 2025We’ve expanded our capabilities with a state-of-the-art, 200,000 sq ft TRC facility in Toronto, completed in 2024 and staffed by over 100 PhD- and MSc-level scientists. This investment enables the development of more innovative compounds, a broader catalogue and custom offering, and streamlined operations for faster delivery. • Our extensive range of over 100,000 high-quality research chemicals—including APIs, metabolites, and impurities in both native and stable isotope-labelled forms—provides essential tools for uncovering molecular disease mechanisms and exploring new opportunities for therapeutic intervention.
New Guide: Characterising Impurity Standards – What Defines “Good Enough?”
April 25th 2025Impurity reference standards (IRSs) are essential for accurately identifying and quantifying impurities in pharmaceutical development and manufacturing. Yet, with limited regulatory guidance on how much characterisation is truly required for different applications, selecting the right standard can be challenging. To help, LGC has developed a new interactive multimedia guide, packed with expert insights to support your decision-making and give you greater confidence when choosing the right IRS for your specific needs.

