On-Line Whole Blood Analysis Using Microextraction by Packed Sorbent and LC–MS-MS
LCGC North America
A new sample preparation method is described for whole blood sample analysis in clinical and preclinical studies.
Microextraction by packed sorbent (MEPS) is a new technique for sample preparation that can be connected on-line with liquid chromatography (LC) or gas chromatography (GC) systems without any modifications. This article describes the use of MEPS in clinical and preclinical studies to quantify different drugs in whole blood samples. MEPS was used to determine cyclophosphamide in mouse blood from preclinical studies using 20 µL of blood samples. The interday accuracies and precisions ranged from 107–109% and from 2.0–7.0%, respectively. The determination of four immunosuppressive drugs in human blood by MEPS and liquid chromatography–mass spectrometry (LC–MS) is described. The method showed a good selectivity and sensitivity. The calibration curves for everolimus, sirolimus, and tacrolimus ranged from 0.5 to 50 ng/mL and for cyclosporine from 3.0 to 1500 ng/mL. Intraday precisions for the studied immunosuppressive drugs were 2.0–11.7% and interday precision ranged from 5.1 to 13.7% (CV).
Off-line sample preparation is a limiting step for achieving high-throughput bioanalysis, indicating a clear need for on-line sample preparation to speed-up the process. Solid-phase extraction (SPE) is attractive for recovering drug residues because of its ability to efficiently retain highly functionalized compounds from aqueous samples and then release them into organic solvents on elution. SPE methods are useful for complex biological samples because the main requirements of the extraction (matrix exchange, desalting, removal of macromolecules and highly polar compounds) are well matched to the properties of the sorbent. When conventional SPE was introduced to the market, it was well accepted by those studying metabolism and toxicology because of the superior efficiency and selectivity that the technique offered over liquid–liquid partitioning methods. Liquid–liquid extraction (LLE) methods require solvent or solvent mixtures of fairly high polarity, and these often yield emulsions and high matrix background in the extracted sample.
For even more-polar analytes, such as drug conjugates and metabolites, recovery and sample purity is further compromised. The same reasons that make conventional SPE so attractive to the study of drug and toxin residues are equally applicable to microextraction by packed sorbent (MEPS) because both are based on the same sorbent chemistries. MEPS uses the same sorbents as conventional SPE columns and so is suitable for use with most existing methods by scaling down the reagent and sample volumes. The excellent performance of MEPS was recently illustrated by on-line liquid chromatography–mass spectrometry (LC–MS) and gas chromatography (GC)–MS assays of drugs and metabolites in water, urine, plasma and whole blood samples (1–39).
In this article, the application of MEPS for the determination of drugs in whole blood samples will be described and the unique characteristics that make MEPS a credible alternative to conventional SPE in this area will be discussed.
The Role of Sample Preparation in Bioanalysis
The matrix problems may call for specific skills in quantitative bioanalysis, particularly in electrospray ionization (ESI) MS detection. Signal suppression caused by unknown matrix interferences is often observed and can give rise to incorrect data interpretation, such as poor accuracy and precision. Therefore, the sample preparation is an important step in bioanalysis processing to avoid ion suppression phenomenon. The sample preparation methods as the first step in an analytical procedure are very important for total analysis processing. If an unsuitable sample preparation method has been used before the injection, the whole analytical process can be wasted. The purpose of the sample preparation is the removal of specific interferences;the conversion of the analytes into a more suitable form for injection, separation and detection; the preconcentration (enrichment) of the analytes to improve the method sensitivity; and to eliminate ion suppression in ESI-MS.
An ideal sample preparation method should involve a minimum number of working steps; be fully automated; and must be highly reproducible with a high recovery of the target analytes. Because of the low concentration levels of drug in plasma and the variety of the metabolites, the selected extraction technique should be virtually exhaustive. Commonly used sample preparation methods in pharmaceutical industry are SPE, LLE, and protein precipitation. The growing number of samples to be analyzed requires high-throughput and fully automated analytical techniques.
Recent developments of sample handling techniques are directed, from one side, toward automation and on-line coupling of sample preparation units and detection systems and from another, toward development of more selective sorbents, such as molecular imprinted polymers (MIPs) and restricted access materials (RAMs). LLE and SPE are the most important sample preparation methods in bioanalysis. SPE methods are effective for complex biological samples because the main requirements of the extraction (matrix exchange, desalting, and removal of macromolecules and highly polar compounds) are well matched to the properties of the sorbent. MEPS is a logical extension of SPE for handling bio-fluids because the small operating volumes reduce the size of sampling. Reducing the demand for sample can extend the use of MEPS to experiments that were previously beyond the scope of SPE (for example, multiple collections of fluid from small animals or volume-limited in-vitro experiments, confirmatory testing of multiple drug residues in hair, saliva, or cerebrospinal fluid) as well as making the sample collection less invasive.
MEPS
MEPS is a new embodiment of SPE that has been miniaturized to work with sample volumes as small as 10 µL and larger (up to 250 µL). MEPS uses the same sorbents as conventional SPE columns, but unlike conventional SPE columns, the MEPS sorbent bed is integrated into a liquid-handling syringe that allows for low-void-volume sample manipulations — either manually or in combination with laboratory robotics. MEPS allows sample processing, extraction, and injection steps to be fully automated as an on-line or at-line sampling or injecting device for GC or LC.
In MEPS, the sorbent, 1–4 mg, is either inserted into the syringe (100–250 µL) barrel as a plug or between the needle and the barrel as a cartridge. The cartridge bed can be packed or coated to provide selective and suitable sampling conditions. Sorbent materials such as silica-based materials (C2, C8, C18), strong cation exchangers using sulfonic acid–bonded silica, RAMs, hydrophilic interaction chromatography (HILIC) carbon, polystyrene-divinylbenzene (PS-DVB) copolymers, or MIPs can be used. MEPS was invented and developed at AstraZeneca in Södertälje, Sweden (1,2).
While the capacity for truly miniaturized extractions allows sparing use of sample and reagents, a more significant strength of MEPS is the ability to fully elute analytes from the sorbent with volumes of 20–50 µL through a syringe needle. Such capability allows MEPS to be adapted for on-line use with both GC and LC inlets or in 96-well plate format for use with immunoassay and other colorimetric techniques.
MEPS Procedures and Extraction Protocols
MEPS extraction steps are the same as for standard SPE: Extraction, washing, and elution have to be optimized to reach the highest analyte recovery. In MEPS extraction procedures, additional steps for postcleaning and re-conditioning have to be included to enable multiple uses of MEPS sorbent. The extraction protocol presented in Figure 1 is intended for C8 and C18 as sorbents and in Figure 2 for mixed-mode sorbents. Blood samples need to be diluted 20–25 times with water or acidic water whereas plasma samples need to be diluted five times. Using a mixed-mode approach (anion–cation exchange) the sample pH must be adjusted to get charged analytes. The first stage is sample loading and can be done once or more depending on sample concentration and if preconcentration of the sample is required. In general four sample-loads (4 ×100 µL) are enough for the preconcentration of analytes and to achieve a good recovery. In addition, the speed of the plunger movement can range from 10 to 20 µL/s to get a high extraction recovery.


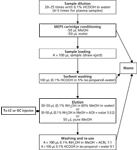
Figure 1: MEPS extraction protocol for blood and plasma samples using C4, C8, or C18 sorbents.
The second stage is a washing step and this is done with 100 µL of a 0.1% HCOOH in 5% isopropanol–water solution (or with 5% methanol in water). The third stage is the elution step and in this step pure methanol can be used (for on-line GC injection) or a mixture of methanol–acetonitrile and water at the correct pH (for reversed-phase LC injection). Furthermore, for mixed-mode MEPS, the pH plays an important role (control charged–uncharged analyte) in achieving a high recovery. In addition, the best possible elution solvent should elute the analyte using the smallest volume of solvent possible. The elution volume can range from 20 to 50 µL and can be eluted in one step (50 µL directly) or by two steps (2 × 25 µL). Finally, a postclean step after the extraction procedures to clean the sorbent, avoid carryover and re-use the MEPS sorbent can be performed by weak and strong washing. The weak washing can consist of water (90%) and small amount of solvent such as methanol or isopropanol (10%). The strong washing can be 0.2% ammonium hydroxide in methanol or acetonitrile — or a mixture of both.

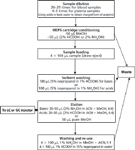
Figure 2: MEPS extraction protocol for blood and plasma samples using mixed-mode sorbent (anionâcation exchange) for acids (pK = 2â8) and bases (pK = 2â10).
A key step for any analytical protocol is the determination of the optimum analyte recovery for the overall method and for each individual procedure. The extraction protocols for C4, C8, and C18 may need adjustments for some analytes that are highly lipophilic or hydrophilic by altering the polarity or pH of used solvents. In these cases the wash and elution steps may need some modifications and tetrahydrofuran or hexane can be used as the elution solution for highly lipophilic compounds.
Comparison of MEPS with Other Extraction Techniques
MEPS differs from SPE because the sorbent bed in MEPS is integrated into a syringe that allows low void volume sample manipulations — either manually or automatically — in combination with an autosampler. Additionally, MEPS uses sparse amounts of sample and reagents, but a more significant strength is the ability to fully elute analytes from the sorbent with volumes of less than 10 µL. MEPS is simple and rapid, does not consume large amounts of solvent compared with SPE and LLE methods, and can achieve the same sensitivity.
Although solid-phase microextraction (SPME) is suitable for on-line use in combination with GC or LC methods, its ability to handle low concentrated analytes in small sample volumes (1–2 mL) is limited. In SPME, the sampling fiber is quite sensitive for the nature of sample matrix. MEPS can be used with complex matrices such as urine, blood, or plasma. Anizan and colleagues (39) evaluated SPME and MEPS as extraction techniques for the analysis of steroid metabolites in urine by GC–MS (39). They found that in this case SPME fiber had a short lifetime. After only five extractions of urine samples, a degradation of the SPME fiber was observed. In addition, they reported that MEPS could be performed rapidly, repeatably, and with a high throughput. They reported that the SPME extraction time was 60 min and the recovery was below 11%, and MEPS recovery was 60–98% with an extraction time of 3 min for all model compounds.
The differences between MEPS and Zip-tips (packed tips, EMD Millipore, Billerica, Massachusetts) is that MEPS can be adapted for on-line use with both GC and LC inlets or in 96-well plate format for use with immunoassay and other colorimetric techniques. Additionally, the MEPS sorbent bed can be used for at least 100 extractions — not just once.
Stir-bar sorptive extraction (SBSE) is regarded as being more difficult to automate, but semiautomated protocols have been described in the literature. SBSE has a good recovery but has a long extraction time of 30 min or more compared with 1–3 min for MEPS.
MEPS Applications for Blood Samples
Determination of Cyclophosphamide in Mouse Blood
Cyclophosphamide is one of the most used anticancer drugs and is used in the treatment of malignant diseases such as leukemia, lymphoma, and solid tumors. In this study MEPS was used to extract cyclophosphamide from mouse blood in a preclinical study (21).
Aliquots (20 µL) of blood samples, standards, and QC samples were diluted with water (1:25). The MEPS sorbent (C2, 1.0 mg) was first conditioned with 50 µL of methanol followed by 50 µL of water. The spiked blood and blood samples were withdrawn into the syringe by the autosampler (4 × 100 µL). The sorbent was then washed once with 100 µL of 95:5 (v/v) water–methanol to remove proteins and other interferences. The analytes were then desorbed by 40 µL 90:10 (v/v) methanol–water directly into the LC injector. Cleaning of the sorbent was carried out using 4 × 250 µL elution solution followed by 4 × 250 µL of the washing solution between every extraction. The same packing bed was used for about 100–120 extractions before it was discarded.
A gradient HPLC method was used. Mobile phase A was 0.1% formic acid in water and acetonitrile (90:10, v/v) and mobile phase B contained 0.1% formic acid in water and acetonitrile (20:80, v/v). The gradient started from 1.0% B up to 50% from 1 to 4 min and then from 4 to 6 min to 90% B and at 6.5 min phase B was set at 1.0% again. All experiments were conducted using a Micromass Ultima triple-quadrupole MS system (Waters Corporation, Manchester, UK). The MEPS syringe and cartridge was obtained from SGE Analytical Science (Melbourne, Australia).

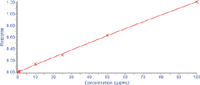
Figure 3: A typical standard curve of cyclophosphamide in whole mouse blood.
Calibration curves of mouse blood samples spiked with cyclophosphamide standards were obtained in the range 0.1–100 µg/mL. The coefficients of determination (r) were >0.99 for all runs. Figure 3 shows a standard curve of the cyclophosphamide in mouse blood using MEPS–LC–MS-MS. The interday mean accuracy was 107–109% using MEPS–LC–MS-MS. The data of interday variation of the precision (CV) were in the 2.0–7.0% range. The data of withinbatch variation of the precision (RSD) were in the 4.0–6.0% range. The accuracy and precision results are summarized in Table I. The carryover was less than 0.4% after the first blank and after the third blank it was negligible. The method was used for the determination of cyclophosphamide in mouse blood from preclinical studies.

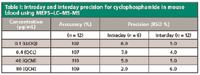
Table I: Intraday and interday precision for cyclophosphamide in mouse blood using MEPSâLCâMS-MS
Determination of Immunosuppressive Drugs in Human Blood
Four immunosuppressive drugs, cyclosporine, everolimus, sirolimus, and tacrolimus in human blood were extracted and determined by MEPS followed by LC–MS-MS (37). In this study, different kinds of sorbent material were investigated and the highest recoveries were obtained with C8 sorbent compared with C18 and C4. Human blood (50 µL) spiked with cyclosporine, everolimus, sirolimus, and tacrolimus was first diluted 25 times with 0.1% formic acid in water and then extracted on a C18 MEPS-cartridge (4 × 100 µL sample loading). The sorbent was then washed once with 100 µL of 5% methanol in water. The analytes were then desorbed by 50 µL 50:30:10:10 methanol–isopropanol–acetonitrile–water. Cleaning of the sorbent was carried out using 4 × 250 µL elution solution (methanol–isopropanol–acetonitrile–water) followed by four 250-µL volumes of the washing solution (5% methanol in 0.1% formic acid) after every extraction. This step decreased memory effects, but also functioned as conditioning step before the next extraction. The same packing bed was used for 100 extractions before it was discarded.
A gradient LC system coupled to a triple-quadrupole mass spectrometer was used, and the total run time for each sample was 2.5 min. The method showed good selectivity for all analytes. The calibration curves for everolimus, sirolimus, and tacrolimus ranged from 0.5 to 50 ng/mL and for cyclosporine from 3.0 to 500 ng/mL. Representative mass chromatograms of blank human blood and spiked human blood with cyclosporine are presented in Figure 4.

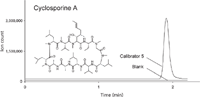
Figure 4: SRM chromatograms obtained from blank human blood and spiked human blood sample with cyclosporine at 500 ng/mL.
Intraday precisions for the studied analytes were 2.0–11.7% (CV) and interday precision ranged from 5.1 to 13.7% (CV). The accuracy values were 102–109%. The mean extraction yield was found to be 97% for cyclosporine, 92% for everolimus, 90% for sirolimus, and 96% for tacrolimus. The method carryover was 0.02% or less.
The MEPS–LC–MS-MS was compared with protein precipitation followed by LC–MS-MS, which was used as a routine method. Patient's samples were analyzed using both methods. The results showed a good agreement between MEPS and protein precipitation but MEPS gave cleaner extracts and no matrix effects were observed. In addition the sensitivity was improved with MEPS as a result of a pre-concentration effect on the MEPS sorbent. MEPS showed better robustness of the chromatography and MS system compared with the protein precipitation method.
Determination of Local Anesthetics in Human Blood
Sample preparation for blood samples is more often concerned with achieving a sufficiently high enrichment of analyte so that detection is possible. In this study (36) human blood (50 µL) spiked with lidocaine, ropivacaine, and bupivacaine was extracted on a C18 MEPS-cartridge conditioned with methanol (50 µL) and water (50 µL). The sorbent was then washed once with 100 µL of water. The analytes were then desorbed by 50 µL of 60:40 (v/v) methanol–water containing 0.25% ammonium hydroxide directly into the injector. Cleaning of the sorbent was carried out using 3 × 250 µL elution solution (60:40 [v/v]methanol–water containing 0.25% ammonium hydroxide) followed by 3 × 250 µL of the washing solution (water) between every extraction. This step decreased memory effects, but also functioned as a conditioning step before the next extraction. The same packing bed was used for about of 50 extractions before it was discarded. A 50 mm × 2.1 mm, 3.5-µm Zorbax SB-C8 column (Agilent Technologies, California) was used as the analytical column and gradient LC was used. Mobile phase A was 0.1% formic acid in acetonitrile and water (10:90) and mobile phase B contained 0.1% formic acid in acetonitrile and water (80:20). The gradient started from 10% B up to 80% B from 1 to 4 min, from 4 to 5 min it was isocratic at 80% B, and at 5.1 min phase B was set at 10% again. The LC was connected to a triplequadrupole MS system.
The calibration curves in human blood were used for the analytes in the range of 10–10,000 nmol/L. The method was validated using 2 H3-lidocaine as the internal standard (IS). Correlation coefficient (r) values obtained were over 0.995. The intraday precision values were 1.0–3.0% for lidocaine, 2.0–4.0% for ropivacaine, and 2.0–5.0% for bupivacaine. The interday precision values were 2.0–7%, 2.0–12%, and 5.0–11% for lidocaine, ropivacaine, and bupivacaine, respectively. The accuracy varied from 90 to 109% for lidocaine, from 85 to 97% for ropivacaine, and from 89 to 97% for bupivacaine (Table II). The accuracy and precision were well in line with the international criteria. The validation of the methodology showed that the method is highly selective for human blood samples.

Table II: Intraday and interday precision for lidocaine, ropivacaine, and bupivacaine in human blood at various concentrations using MEPSâLCâMS-MS
The extraction recovery was about 90% for all analytes (MEPS sampling, 3 × 100 µL). Figure 5 shows a mass chromatogram of direct injection of lidocaine, ropivacaine, and bupivacaine in the mobile phase, and a blood sample after MEPS extraction.

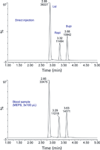
Figure 5: Comparison of mass chromatograms of lidocaine (1000 nmol/L), ropivacaine (500 nmol/L), and bupivacaine (500 nmol/L) obtained by direct injection (in the mobile phase) and after extraction from spiked human blood by MEPS.
In this study, the carryover was investigated by injecting elution solution after the highest standard concentration; the carryover was about 0.02% after washing five times with methanol containing 0.2% ammonium hydroxide and five times with water.
Determination of a New Pro-Drug: Neuronal Nicotinic Receptor Partial Agonist in Human Blood
A neuronal nicotinic receptor (α4β2 NNR) agonist for treatment of cognitive deficits in Alzheimer's disease was determined in human blood by MEPS–LC–MS-MS (38).
A mixed-mode sorbent (polystyrene ENV+) and silica-based ion-exchange (strong cation exchange) sorbent were used. Aliquots (20 µL) of blood samples were diluted 25 times with 0.1 M ammonium formate. The MEPS sorbent was washed first with 100 µL 0.1 M ammonium formate. A 100-µL volume of diluted blood was loaded and the sorbent was then washed once with water (100 µL). The pro-drug was desorbed by 50 µL of 60:40 (v/v) methanol–water containing 3% ammonium hydroxide and then 3 µL was injected into an ultrahigh-pressure liquid chromatography (UHPLC) injector that was coupled to a Xevo-QT mass spectrometer (Waters, Milford, Massachusetts). The separation was performed on an 50 mm × 2.1 mm, 1.7-µm Acquity C18 column (Waters).
The calibration curve in human blood was used for the pro-drug in the range of 2.0–5800 nmol/L. Correlation coefficient (r) values obtained were over 0.999. Good accuracy and precision for the QC samples at three different concentrations levels were obtained. The LLOQ was set to 2.0 nmol/L. Figure 6 shows a chromatogram with MS detection at LLOQ in human blood. MEPS was compared with the protein precipitation for blood samples, ion suppression was observed, and bad linearity was obtained using protein precipitation instead of MEPS.

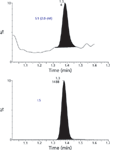
Figure 6: MRM mass chromatogram at LLOQ (2 nmol/L) of a new pro-drug (neuronal nicotinic receptor agonist) obtained from spiked human blood.
Conclusion
MEPS is a miniaturized presentation of SPE that uses 10–100 times less sample and reagents than commercial SPE. The same sorbent can be used for more than 100 extractions for blood and plasma samples. The small volumes also significantly reduce extraction time from 10–15 min down to 1–2 min. The MEPS format allows routine SPE of samples at 10 µL and up to 250 µL in a single cycle. Very small elution volumes and the integration of the sorbent into the sampling syringe and needle allow MEPS to be used for direct injection into LC and GC systems. The format is also suitable for complete automation with on-line sample handling robots and autosamplers. The number of reuses possible for a MEPS cartridge is dependent on the sample matrix type. As with conventional SPE, maintaining functional flow during the lifetime of the device is matrix dependent.
References
(1) M. Abdel-Rehim, AstraZeneca Application, Current Patents Gazette, week 0310, WO 03019149, 77 (2003).
(2) M. Abdel-Rehim, PCT, World Intellectual Property Organization, International Publication Number: WO 03/019149 A1, International Publication Date: 6 March 2003.
(3) M. Abdel-Rehim, J. Chromatogr. B 801, 317–321 (2004).
(4) M. Abdel-Rehim, Z. Altun, and L. Blomberg, J. Mass Spectr. 39, 1488–1493 (2004).
(5) Z. Altun, M. Abdel-Rehim, and L.G. Blomberg, J. Chromatogr. B 813, 129–135 (2004).
(6) M. Vita et al., J. Chromatogr. B 817, 303–307 (2005).
(7) M. Vita et al., J. Chromatogr. B 821, 75–80 (2005).
(8) M. Abdel-Rehim et al., Anal. Chim. Acta. 539, 35–39 (2005).
(9) Z. Altun et al., J. Liq. Chromatogr. & Relat. Technol. 29, 829–839 (2006).
(10) M. Abdel-Rehim et al., J. Liq. Chromatogr. & Relat. Technol. 29, 1725–1736 (2006).
(11) M. Abdel-Rehim, M. Dahlgren, and L. Blomberg, J. Sep. Sci. 29, 1658–1661 (2006).
(12) M. Abdel-Rehim et al., J. Liq. Chromatogr. & Relat. Technol. 29, 2537–2544 (2006).
(13) M. Abdel-Rehim et al., J. Liq. Chromatogr. & Relat. Technol. 29, 2413–2424 (2006).
(14) A. El-Beqqali, A. Kussak, and M. Abdel-Rehim, J. Chromatogr. A 1114, 234–238 (2006).
(15) A. El-Beqqali et al., J. Liq. Chromatogr. & Relat. Technol. 30, 575–586 (2007).
(16) A. El-Beqqali, A. Kussak, and M. Abdel-Rehim, J. Sep. Sci. 30, 421–424 (2007).
(17) A. El-Beqqali and M. Abdel-Rehim, J. Sep. Sci. 30, 2501–2505 (2007).
(18) M. Abdel-Rehim et al., J. Liq. Chromatogr. & Relat. Technol. 30, 3029–3041 (2007).
(19) R. Said et al., J. Liq. Chromatogr. & Relat. Technol. 31, 683–694 (2008).
(20) M. Abdel-Rehim et al., J. Chromatogr. Sci. 46, 518–523 (2008).
(21) M. Kamel et al., The Open Spectroscopy Journal 3, 26–30 (2009).
(22) Z. Altun and M. Abdel-Rehim, Anal. Chemica Acta. 630, 116–123 (2008).
(23) H. Miyaguchi et al., J. Chromatogr. A 216, 4063–4070 (2009).
(24) S. Matysik and F.-M. Matysik, Microchim Acta 166, 109–114 (2009).
(25) Application Note, SGE Analytical Science, http://www.sge.com/
(26) G. Morales-Cid et al., Anal. Chem. 81, 3188–3193 (2009).
(27) G. Morales-Cid et al., Electrophoresis 30, 1684, (2009).
(28) E. Jagerdeo and M. Abdel-Rehim, J. Am. Soc. Mass Spectrom. 20, 891–899 (2009).
(29) H. Miyaguchi et al., J. Chromatogr. A 1216, 4063–4070 (2009).
(30) S. Matysik and F.-M. Matysik, Microchim Acta 166, 109–114 (2009).
(31) Application Note, SGE Analytical Science, http://www.sge.com.
(32) G. Morales-Cid et al., Anal.Chem. 81, 3188–3193 (2009).
(33) G. Morales-Cid et al., Electrophoresis 30, 1684 (2009).
(34) M. Abdel-Rehim, LCGC Europe 22(1) (2009).
(35) M. Abdel-Rehim, J. Chromatogr. A 1217, 2569–2580 (2010).
(36) R. Said et al., Bioanalysis 2(2), 197–205 (2010).
(37) R. Said et al., submitted to Therapeutic Drug Monitoring.
(38) M. Abdel-Rehim, in manuscript.
(39) S. Anizan et al., J. Chromatogr. A 1217, 6652–6660 (2010).
Mohamed Abdel-Rehim specializes in sample preparation and the analysis of drugs in biological samples by LC–MS. He currently works at AstraZeneca (Global DMPK), Södertälje, Sweden. He is a professor in the Department of Chemistry and Biomedical Sciences at Karlstad University and in the Department of Analytical Chemistry at Stockholm University, Sweden. Additionally, he is a visiting professor at the National Research Center of Egypt. The main focus of his research is the development of highly automated and integrated instrumentation for the isolation of drugs and metabolites from complex matrices and the subsequent separation, identification and determination of these drugs using GC–MS and LC–MS–MS. He is also interested in the preparation of new sorbents such as monolithic phases for applications in bioanalysis.


















Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).




Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.

