Mass Spectrometry of Organic Molecules and Laser-Induced Acoustic Desorption: Applications, Mechanisms, and Perspectives
This study illustrates the versatility of a laser-induced acoustic desorption (LIAD) approach for volatilization of analytes in modern mass spectrometry.
The usefulness of a laser-induced acoustic desorption (LIAD) method for analytical mass spectrometry (MS) of organics is a well-established fact. However, the mechanism of LIAD is not well-understood, which limits widening of its applications in practical MS. Aiming at clarification of the mechanism, key physical parameters of desorbed molecules, such as their energy and velocity distributions, have been measured in this work. Our own study, as well as the analysis of existing literature data on LIAD, led us to a conclusion that this phenomenon could not be explained in the framework of a simple "shake off" mechanism and required a more sophisticated model. For the most often used experimental arrangement, we have demonstrated that the desorption process is synergistically driven by distortion and heating of the analyte film followed by its cracking and formation of nonequilibrium "desorption sites" on the cracks' edges resulting in volatilization of the analyte molecules. At the same time, under different experimental conditions (for example, high driving laser fluences or the use of confined targets), real shock waves may be generated in the substrate and the "shake off" mechanism of desorption could not be excluded, especially for nano- and bio-particles. This study illustrates the versatility of an LIAD approach for volatilization of analytes in modern MS.
The ability of lasers to concentrate significant energy in small volumes within a short time period has resulted in a long lasting and strong interest in the mass-spectrometry (MS) community. In many MS applications, lasers are used for both volatilization of analytes and for ionization of the analyte species in the gas phase before passing them through mass analyzers (1). Applied to MS analyses of complex organics, this approach has a very significant drawback: the high-density hot laser plasma created during the analyte irradiation causes molecular fragmentation due to extensive atomic and molecular collisions in the plasma, and thus key information on the analyte molecular composition is lost. This is why the search for new, gentler methods for the molecules' evaporation and volatilization and softer methods for their consequent ionization remains a challenge for laser-based MS.
One brilliant solution resulting from such search efforts was the matrix-assisted laser desorption and ionization (MALDI) method, which became extremely popular in practical MS analyses of organics, to the extent that the Nobel Prize in Chemistry was awarded to its inventor in 2002 (2,3). Presently, MALDI is considered one of the most powerful and informative tools for MS analysis of organics. It has become, in fact, the industry standard. However, despite its great success, many inherent drawbacks of MALDI prevent it from becoming the absolute and only choice for MS analysis of organics (4). And the search for alternative MS methods using lasers for volatilization and ionization of the molecules is ongoing.
Ironically, in the same year that the first observation of MALDI was reported (1985) (5), the other phenomenon, later called laser-induced acoustic desorption (LIAD), was discovered by B. Lindner and U. Seydel. In their pioneering work (6), they reported the observation of emission of ions of organic molecules deposited on the front surface of a thin copper foil whose backside was irradiated by laser pulses. In the shadow of the spread of MALDI, this observation did not get the attention it deserved until the mid-1990s when C.H. Chen's team (7) published its own observation of the emission of charged particles (ions and electrons) under back-side laser irradiation conditions and proposed the physical mechanism for this phenomenon, as well as its name and the acronym LIAD. According to their hypothesis, the emission–desorption from the front surface of back-irradiated substrate was caused by the generation of strong acoustic waves in its material resulting from the interaction between the backside surface and the laser plasma plume. These acoustic waves penetrating from the backside toward the front surface initiate its reciprocal motion with the velocity close to the speed of sound and simply "shake off" the surface analyte molecules similar to someone shaking off dust particles from their clothes.
This "shake off" idea quickly became the dominant hypothesis regarding the nature of LIAD and facilitated many successful LIAD experiments with different analyte species. Unfortunately, the "first-principles" physical analysis of the "shake-off" mechanism casts serious doubts about its validity, and particularly its ability to initiate molecular desorption. To be "shaken off" the surface, the molecules must have the normal component of their velocity sufficient to overcome the surface energy barrier of εb~0.05–0.5 eV (8) (the binding energy of physically adsorbed molecules):


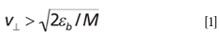
in which M is the mass of the molecule. For a range of moderate masses (~500), one can estimate that

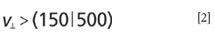
in units of meters per second. These values are comparable with speed of sound in solids, but it is a common mistake to associate the speed of sound with the velocity of mass transfer in acoustic vibrations, which in reality is much lower (9) and, in most cases, insufficient for successful molecular desorption. This is why the "shake off" hypothesis does not work well as the explanation of the LIAD mechanism.
Experiments on LIAD
A much wider application of LIAD to MS analysis of a broad range of organic species started with the work of H. Kenttamaa and colleagues (10). A typical arrangement of LIAD MS experiments is demonstrated in Figure 1. The analyte is applied to one (front) side of a metal foil (6–15 µm thick) whereas the backside is subjected to laser pulses (typical pulse duration 10–20 ns, and power density is in the range of 0.1–1 GW/cm2 ). Because the desorbed molecules are neutral, for further MS analysis they must be post-ionized in some way. In Figure 1, a single-photon ionization (SPI) method is demonstrated, but other approaches, such as electrospray ionization (ESI), chemical ionization (CI), and electron ionization (EI) are also widely used (2). One of the key advantages of LIAD is that it can be easily coupled with different kinds of MS instrumentation, both of commercial and home-made designs. Figure 1 illustrates the use of time-of-flight (TOF) MS, but other types of MS instruments have also been used for LIAD studies, for example linear-quadruple ion-trap (LQIT) and Fourier-transform cyclotron resonance (FT-ICR) mass spectrometers.

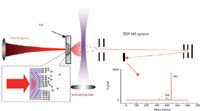
Figure 1: Schematic drawing of a typical LIAD experimental arrangement.
In general, the results of LIAD applications to the analysis of different organic species are very encouraging, if not spectacular. In their first work, H. Kenttamaa and colleagues (10) reported the desorption of a wide variety of organic compounds in the mass range of 110–2500 amu. Successful volatilization of intact, thermally labile molecules was reported. Further LIAD studies expanded the applicability of LIAD on other groups of organic and biomolecules such as alanylalanine and thymidine (11), dinucleoside phosphates (12), saturated hydrocarbons (13), tetrapeptides (14), organic dyes (15), and even bioparticles or viruses (16). The unique feature of LIAD is the ability to volatilize mixtures of different organics, which was demonstrated by the MS analyses of petroleum distillates (17), asphaltenes (18), and petroleum model compounds (19). This feature makes LIAD suitable for MS analysis of mixtures of chemicals with unknown composition, which is extremely difficult to accomplish by other MS methods, such as MALDI.
Besides the demonstration of the ability of LIAD to desorb intact molecules, a few attempts to study its physical nature were done. An experimental study of internal energies of the molecules volatilized by LIAD demonstrated that this approach does not noticeably increase the internal energy of the desorbed molecules (20), observed minimal fragmentation of the molecules, and suggested that thermal processes have minor influence on the physics of LIAD.
Interesting results were obtained in studies of how the signal of molecules desorbed by LIAD depends on the driving laser intensity and on the number of laser shots striking the same spot. It was found that the signal increases with the increase of laser intensity (14) and that this increase is described by the exponential law (15). However there is the limit of applied laser intensity that is defined by the resistance of the given foil to laser damage. It was experimentally found for Ta and Ti foils that the thickness of about 12 µm is close to optimal (14,15,20). Another LIAD feature observed was that the desorption signal did not noticeably decrease after a single laser shot, and instead it remained significant after thousands of such shots, although its magnitude gradually decreased (14,15). It was found that this decrease obeyed the power law, instead of the exponential one (15). This supports the hypothesis of multiple independent desorption sites on the surface (each one obeying the exponential law) and contradicts the model of a simple exhaust of analyte amount on the surface (15).
In general, despite numerous successful applications and rather intensive fundamental studies, the LIAD approach has not yet become a widespread method such as MALDI. In our opinion, there are two main reasons for this. The first one is, apparently, the need for an ionization step, which makes the LIAD arrangement somewhat more complicated. The second one is the lack of the adequate physical mechanism; we will try to discuss this problem in more detail.

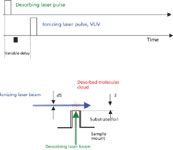
Figure 2: The scheme of experimental arrangement of TOF technique for the measurement of energy and velocity distributions of neutral molecules.
Energy Spectra of Desorbed Molecules
Even in early works on LIAD, two distinctive features of the physical mechanism were pointed out: the desorption of thermally labile molecules in their intact form, and the very small velocities of desorbed molecules (10). These observations clearly indicate the invalidity of other hypotheses assuming thermal origins of LIAD. At the same time, the "shake-off" mechanism by acoustic waves also is very unlikely. Therefore, the mechanisms of energy transfer between the laser pulse and the adsorbed molecules remains unclear, and more information about physical characteristics of the molecular desorption process needs to be obtained. The most informative characteristic of desorbed molecules is their velocity (or energy) distribution. For neutral particles that need to be post-ionized, such as molecules desorbed by LIAD, the most common approach to measure it is the use of the TOF technique proposed by F. Hillenkamp (21). The experimental arrangement (15) is schematically shown in Figure 2. The LIAD driving laser pulse (10-ns duration, 1–5 mJ/pulse energy) strikes the backside of the foil at the moment in time t = 0. The ionizing laser beam (10-ns pulse duration, 2-mJ/pulse energy) intercepts the volatilized molecular cloud at the distance S above the foil surface. The delay time between the driving and ionizing laser pulses t can be varied from pulse to pulse and, therefore signal-vs.-delay dependencies can be obtained. Such a dependence for rhodamine B is presented in Figure 3. For comparison, the same dependence obtained for conventional laser desorption (LD) (for example, with direct laser irradiation of the front surface of the target) under exactly the same experimental conditions is also plotted in Figure 3. The main difference between these two plots is apparent: LIAD-desorbed molecules are much slower than those volatilized by LD. To get the energy distribution from the signal-vs.-delay dependence, one needs to take into account that the Jacobian of this variable transform given by S2/t3 (22). Obtained by such a procedure, energy distributions for organic dye molecules, rhodamine B, fluorescein, methylanthracene (MA), coumarin-522 (N-methyl-4-trifluoromethylpiperidino[3,2-g]coumarin), and BBQ (4,4"-bisbutyl octyloxy-p-quaterphenyl), are plotted in Figure 4. The mean energies of molecules in LIAD experiments (as well as their mean velocities) showed no apparent trend with the increase of desorbing laser fluences maintaining the average value at about 9 meV for rhodamine B, 9.5 meV for BBQ, 6.5 meV for coumarine 522, and 7.5 meV for methylanthracene. The dashed line represents Maxwell distribution for rhodamine B corresponding to the temperature of 80 K. This physically unrealistic temperature was chosen to obtain the best fit at the distribution maximums. Also, high energy tails of the experimental energy distributions strongly deviate from the exponential law, which is apparent on the double logarithmic scale of plots in Figure 4 and reveal behavior closer to the power dependence.

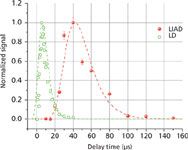
Figure 3: Signal-vs.-delay dependencies for LIAD and LD desorbed rhodamine B molecules.
Mechanisms of LIAD
The results on kinetic energy distributions of desorbed molecules mentioned above suggest that the conditions of the LIAD process are far from equilibrium and that there is no single simple mechanism of energy transfer in LIAD. The literature analysis of information on emission and desorption phenomena reveals that there is another desorption process with similar energy spectra. It is called electron-stimulated desorption (ESD) where desorbed particles also have low translational energies and nonequilibrium energy distributions (23). The primary mechanism of ESD is supposed to be the formation of repulsive states on the surface due to electronic excitations as the result of the electron bombardment of the surface. In the case of LIAD, because the foil is opaque, the laser radiation striking the backside cannot be the direct cause of the electronic excitation on the front side surface. However, another mechanism of desorption sites formation first proposed by A. Vertes (24) for the explanation of MALDI might be applicable for LIAD. This hypothesis assumes that the heat generated by the laser pulse in metal can cause a mechanical stress in the thin organic analyte film on the substrate surface due to the mismatch of mechanical and thermal characteristics of the metal substrate and the film. It is hard to verify the validity of such a mechanism in the case of MALDI because of its interference with other phenomena on the surface occurring under strong laser irradiation. In the case of LIAD, when direct interaction between the laser beam and the studied material is excluded, this mechanism might play the dominant role and explain many observed regularities. Another, indirect suggestion of the validity of such a desorption mechanism is the triboemission or the "Kramer effect" (25,26). In essence, the emission of charged particles and photons can be initiated by the surface distortion, such as mechanical deformation, scratching, and bending and does not have to be connected with the thermal excitation.
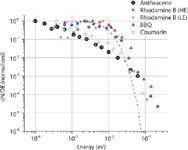
Figure 4: Energy spectra of various organic dye molecules. HE and LE denote rhodamine B spectra obtained at a driving laser fluency of 3.4 J/cm2 and 2.3 J/cm2, respectively. The dashed curve is the equilibrium Maxwell-Boltzmann distribution at 250 K.
It is a well-known fact that a film deposition on a substrate surface results in many cases in the residual mechanical stress (due to the lattice parameters mismatch between the film and the substrate), and thus some excessive potential energy can be stored in the film. This stress can be produced in two ways: one is the growth stress and the other is the induced (or extrinsic) stress (27). An external impact, such as acoustic or thermal wave generated by the laser irradiation of the substrate, should initiate the reconstruction of the film, which can result in releasing this excess energy or, possibly, generating an additional extrinsic stress. In both cases, it can cause formation of cracks in the film so that intermolecular bonds at the edges of the cracks break. As a result of this crack formation process, excited electronic states can form at the crack edges and induce desorption of the molecules. This process is schematically illustrated on the insertion of Figure 1.
It must be emphasized that the process described above is not the only mechanism of LIAD. This is because the acronym LIAD combines a group of different physical phenomena occurring under similar experimental arrangements (volatilization of some species from the front side whereas the backside is irradiated by lasers) but having different natures. Moreover, often somewhat confusing terminology is used. For example, in some LIAD papers, "acoustic wave" is synonymized with "shock wave." In reality, these are very different phenomena with different key manifestations and characteristics. Shock waves in solids can be initiated by very strong laser pulses with intensities exceeding tens of gigawatts per centimeter squared, and they have no material transfer velocity limitations in contrast to acoustic waves (28). Therefore for shock waves, the direct "shake-off" mechanism of adsorbed particles volatilization is very likely, and this is what could drive the desorption of micrometer-sized particles (29). Early observations of biocells and viruses volatilization (16) as well as the emission of charged particles (7) could also be the result of shock wave formation, which was accompanied by significant surface disruption at the micrometer scale.
However, under most common experimental conditions of LIAD, when 10–20 µm thick foils are used, the generation of shock waves is unrealistic. The increase of laser intensities up to tens of gigawatts per centimeter squared will immediately cause damage to (burn through) the foil, formation of a hot plasma plume on the front surface, and strong fragmentation of desorbed analyte molecules.
Future Applications
In our opinion, the LIAD approach has great potential for becoming a mainstream method in the future of analytical MS. Its ability to volatilize a wide variety of molecules (small and macro, deposited on substrates as single species and as mixtures) in intact, nonfragmented form, as well as possible solvent-free and matrix-free sample preparation procedures make it the method of choice in some specific applications. Even the need for postionization does not compromise the advantages of LIAD. The recent successful application of LIAD at ambient air pressure (19,30) demonstrated in combination with ion-mobility MS (31) opens the way to design a vacuum-free analytical MS instrument. The other significant advantage of LIAD is its tolerance for the type of driving laser such that only the peak power of the laser beam is the significant parameter. The recent progress in design and development of all solid-state, robust, and relatively cheap DPSS lasers can make LIAD-based MS instruments a very strong competitor to existing mobile mass-analyzers for express analysis.
An interesting and promising combination of LIAD and chromatography techniques was demonstrated (30) in which a thin-layer chromatography (TLC) plate was used as a substrate. Successful separation of a dye standard mixture on TLC and further mass-analysis with LIAD demonstrated the cheap and effective analysis of a chemical compound mixture.
Conclusion
It is interesting that the acoustic waves that gave the name to LIAD in many experimental arrangements are not the direct trigger of the molecular desorption and volatilization. Instead, a multistep and complex mechanism of energy transfer from the laser beam to adsorbed molecules is likely employed here. Unfortunately, at the moment it is only a hypothesis, which has no direct experimental proofs. To get them and to make LIAD a widespread method in analytical MS, a new series of experimental studies is required.
Another direction for future research in LIAD is in studies of surface conditions and their influence on LIAD behavior. Currently, simple untreated metal surfaces have been employed as substrates for analyte films. No attempts have been made to change the probability of electron exchange between the surface and the outgoing molecules. Pure, cold, and untreated metal surfaces are unlikely to enhance the ionization probability of desorbed particles. On the other hand it is well-known that surface-assisted laser desorption and ionization (SALDI) (32) is the alternative for MALDI. Therefore the search of surface conditions for LIAD that will allow direct desorption of ionized molecules and omit the ionization process is another important direction for future studies.
With all that said, even at its present state of development, LIAD is a very promising and powerful tool for analytical MS applications.
Acknowledgment
This work is supported by the U.S. Department of Energy, BES-Materials Sciences, under Contract DE-AC02-06CH11357, by UChicago Argonne, LLC.
Alexander Zinovev and Igor Veryovkin are with Argonne National Laboratory, Argonne, Illinois.
References
(1) J.C. Miller and R.F. Haglund, Laser Ablation and Desorption. Experimental Methods in the Physical Sciences, (Academic Press, San Diego, California, 1998).
(2) C. Dass, Fundamentals of Contemporary Mass Spectrometry. Wiley-Interscience Series on Mass Spectrometry. (Wiley-Interscience, Hoboken, New Jersey, 2007).
(3) R.B. Cole, Electrospray and MALDI Mass Spectrometry: Fundamentals, Instrumentation, Practicalities, and Biological Applications. 2nd ed. (Wiley-Interscience, Hoboken, New Jersey, 2010).
(4) M.W. Duncan, H. Roder, and S.W. Hunsucker, Briefings in Functional Genomics & Proteomics 7(5), 355–370 (2008).
(5) M. Karas, D. Bachmann, and F. Hillenkamp, Anal. Chem. 57(14), 2935–2939 (1985).
(6) B. Lindner and U. Seydel, Anal. Chem. 57(4), 895–899 (1985).
(7) V.V. Golovlev, S.L. Allman, W.R. Garrett, and C.H. Chen, Applied Physics Letters 71(6), 852–854 (1997).
(8) A.W. Adamson and A.P. Gast, Physical Chemistry of Surface (John Wiley & Sons, Inc., New York 1997).
(9) L.D. Landau and E.M. Lifshits, Fluid Mechanics (Pergamon, New York, 1987).
(10) J. Perez, L.E. Ramirez-Arizmendi, C.J. Petzold, L.P. Guler, E.D. Nelson, and H.I. Kenttamaa, Int. J. Mass Spectrom. 198(3), 173–188 (2000).
(11) C.J. Petzold, L.E. Ramirez-Arizmendi, J.L. Heidbrink, J. Perez, and H.I. Kenttamaa, J. of the Amer. Soc. for Mass Spectrom. 13(2), 192–194 (2002).
(12) J.A. Liu, C.J. Petzold, L.E. Ramirez-Arizmendi, J. Perez, and H. Kenttamaa, J. of the Amer. Soc. for Mass Spectrom. 127(37), 12758–12759 (2005).
(13) J.L. Campbell, K.E. Crawford, and H.I. Kenttamaa, Anal. Chem. 76(4), 959–963 (2004).
(14) R.C. Shea, C.J. Petzold, J.L. Campbell, S. Li, D.J. Aaserud, and H.I. Kenttamaa, Anal. Chem. 78(17), 6133–6139 (2006).
(15) A.V. Zinovev, I.V. Veryovkin, J.F. Moore, and M.J. Pellin, Anal. Chem. 79(21), 8232–8241 (2007).
(16) W.P. Peng, Y.C. Yang, M.W. Kang, Y.K. Tzeng, Z.X. Nie, H.C. Chang, W. Chang, and C.H. Chen, Angewandte Chemie Int. Edition 45(9), 1423–1426 (2006).
(17) K.E. Crawford, J.L. Campbell, M.N. Fiddler, P. Duan, K. Qian, M.L. Gorbaty, and H.I. Kenttamaa, Anal. Chem. 77(24), 7916–7923 (2005).
(18) D.S. Pinkston, P. Duan, V.A. Gallardo, S.C. Habicht, X.L. Tan, K.N. Qian, M. Gray, K. Mullen, and H.I. Kenttamaa, Energy & Fuels 23, 5564–5570 (2009).
(19) L. Nyadong, A.M. McKenna, C.L. Hendrickson, R.P. Rodgers, and A.G. Marshall, Anal. Chem. 83(5), 1616–1623 (2011).
(20) R.C. Shea, C.J. Petzold, J.A. Liu, and H.I. Kenttamaa, Anal. Chem. 79(5), 1825–1832 (2007).
(21) B. Spengler, U. Bahr, M. Karas, and F. Hillenkamp, Anal. Instrum. 17(1–2), 173–193 (1988).
(22) F. Balzer, R. Gerlach, J.R. Manson, and H.-G. Rubahn, J. Chem. Phys. 106(19), 7995–8012 (1997).
(23) C.E. Young, J.E. Whitten, M.J. Pellin, D.M. Gruen, R.L. Benbow, and P.L. Jones, Surface and Interface Analysis 14(10), 647–655 (1989).
(24) A. Vertes in Methods and Mechanisms for Producing Ions from Large Moleculres, K.G. Standing and W. Ens, Eds. (Plenum Press, New York, 1991).
(25) K. Nakayama, N. Suzuki, and H. Hashimoto, J. Phys. D: Appl. Phys. 25(2), 303–308 (1992).
(26) L. Oster, V. Yaskolko, and J. Haddad, Physica Status Solidi a-Applied Research 187(2), 481–485 (2001).
(27) L.B. Freund and S. Suresh, Thin Film Materials. Stress, Defect Formation and Surface Evolution (Cambridge University Press, Cambridge, 2003).
(28) R. Menikoff in Shock Wave Science and Technology Reference Library, Y. Horie, Ed. (Springer, Berlin, 2007), p. <1-3>.
(29) V. Menezes, K. Takayama, T. Ohki, and J. Gopalan, Applied Physics Letters 87(16) (2005).
(30) S.C. Cheng, M.Z. Huang, and J. Shiea, Anal. Chem. 81(22), 9274–9281 (2009).
(31) C.L. Wilkins and S. Trimpin, Ion Mobility Spectrometry-Mass Spectrometry: Theory and Applications (CRC Press, Boca Raton, Florida, 2011).
(32) J. Sunner, E. Dratz, and Y.C. Chen, Anal. Chem. 67(23), 4335–4342 (1995).
Alexander Zinovev and Igor Veryovkin are with Argonne National Laboratory, Argonne, Illinois.

























New TRC Facility Accelerates Innovation and Delivery
April 25th 2025We’ve expanded our capabilities with a state-of-the-art, 200,000 sq ft TRC facility in Toronto, completed in 2024 and staffed by over 100 PhD- and MSc-level scientists. This investment enables the development of more innovative compounds, a broader catalogue and custom offering, and streamlined operations for faster delivery. • Our extensive range of over 100,000 high-quality research chemicals—including APIs, metabolites, and impurities in both native and stable isotope-labelled forms—provides essential tools for uncovering molecular disease mechanisms and exploring new opportunities for therapeutic intervention.
New Guide: Characterising Impurity Standards – What Defines “Good Enough?”
April 25th 2025Impurity reference standards (IRSs) are essential for accurately identifying and quantifying impurities in pharmaceutical development and manufacturing. Yet, with limited regulatory guidance on how much characterisation is truly required for different applications, selecting the right standard can be challenging. To help, LGC has developed a new interactive multimedia guide, packed with expert insights to support your decision-making and give you greater confidence when choosing the right IRS for your specific needs.

