Buffer Preparation — Hints, Tips and Common Errors
LCGC Asia Pacific
Accurate preparation and correct selection of buffers is essential to obtain reproducible and consistent results in capillary electrophoresis (CE). A number of factors should be considered in buffer optimization, including the pKa of the buffering ion and the analyte as well as the molarity of the acid or base used in the adjusting procedure. Accurate recording of the precise reagents used and the procedures performed is necessary to prepare buffers consistently.
Accurate preparation and correct selection of buffers is essential to obtain reproducible and consistent results in capillary electrophoresis (CE). A number of factors should be considered in buffer optimization, including the pKa of the buffering ion and the analyte as well as the molarity of the acid or base used in the adjusting procedure. Accurate recording of the precise reagents used and the procedures performed is necessary to prepare buffers consistently.
"Poor reproducibility of results and poor quantitative precision will be attainable in CE assays without significant attention being paid to the preparation of buffers used."
In this instalment of "CE Currents" buffer selection, buffer preparation and a selection of problems encountered when preparing working buffers for capillary electrophoresis (CE) will be examined. Variation in the buffer preparation in CE has more pronounced effects on the analyte separation than HPLC, therefore, analysts must take great care when describing buffer preparation and following the preparation instructions.
Why Use a Buffer?
The primary role of the buffer is to generate and maintain a set pH because this affects solute ionization and the level of electrosmotic flow (EOF) generated in the capillary. The ionic strength of the buffer also influences the solute migration times and the level of current and EOF generated in the capillary. The term electrolyte is often used in CE. This simply refers to a solution of ions but does not indicate any buffering ability. Failure to use a buffered electrolyte can result in severe operational difficulties because the method will be significantly less robust than a buffered electrolyte.
Electrolytic changes — termed "buffer depletion" — of the buffer can occur, especially in prolonged injection sequences. This results in gradual changes of buffer pH in the separation buffer vials, which leads to changes in migration times and/or separation selectivity. If the electrolyte used has a good buffering capacity then it can actively resist these pH changes.
How Do you Prepare a Buffer?
Let's look at a common buffer described in CE literature — "25 mM phosphate pH 7.0". How exactly is that prepared? From this simple description it is impossible to tell the exact composition and ionic strength and is, therefore, impossible to reproduce.
Table 1 shows how a buffer described in the literature as "25 mM phosphate pH 7.0" could have been prepared: the ionic strengths, buffering capacity, EOF flow-rates and currents (the internal heating in the capillary) will be different for each preparation variant.



Table 1: Variants on buffer preparation procedure.
"The exact procedure for electrolyte preparation should be defined in the method including the electrolyte salt form used and, if used, an exact pH adjustment procedure. The pH adjustment procedure should include details of the nature and concentration of the solution used to adjust the pH of the electrolyte."
Which Buffer Should I Select?
The suitability of a buffer system depends upon many factors, the first being the pKa value of the buffer acid or base. Other factors that should be taken into account include the solubility and stability of the analytes in the electrolyte, the effect of temperature and heat generation. A selection of commonly used buffers are listed in Table 2.

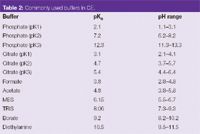
Table 2: Commonly used buffers in CE.
"For a buffer to be effective its pH must be centred around its pKa ± 1 — buffers should be selected accordingly."
The biological buffers or "good buffers" such as tris(hydroxymethyl)aminomethane (TRIS) or 2–(N–morpholino)ethanesulphonic acid (MES) (Figure 1) are widely used in CE because these can be used at much higher concentrations than inorganic electrolytes as a result of their lower conductivities.1 These buffers contain both acid and base functionalities and, therefore, have a positive and negative charge when used exactly at their pKa values. The net zero charge on the buffer species means that they do not generate a current when a voltage is applied, allowing high concentrations to be used with good buffering capacity.

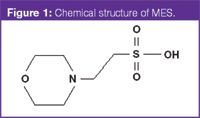
Figure 1
What About Buffer Counter-Ions?
The counter-ion of a buffer also migrates when a voltage is applied and, therefore, generates current. It may be possible to reduce the current by switching to a counter-ion with a smaller ionic radius. Three different ionic radii phosphate buffers were investigated under the same operating conditions, at the same concentration (50 mM) and at the same pH (7.5).
The test mix used was methimazole, warfarin, prenidisolone and naphthoxy acetic acid at 100 μg/mL dissolved in water. The data in Table 3 illustrates that the larger the counter-ion, the higher the current which is generated and the longer the solute migration time.2


Table 3: Migration times for naphthoxy acetic acid, current and ionic radius data for three different phosphate buffers.
"It is important when quoting methods, or repeating other analysts work, to closely observe the counter-ions used. This is also the situation when adjusting the buffer pH in the buffer preparation phase."
The selection of the buffer and counter-ion also affects the symmetry of the separated peaks. The buffer ions and counter-ions migrate along the capillary when the voltage is applied. This is what generates the current in the capillary. If the migration speed of the peaks and the counter-ions is very different then a process called electrodispersion occurs, leading to peak distortion. For example, the selection of a sodium phosphate buffer can lead to peak distortion for basic drugs so a 0.01M hydrogen phosphate and 0.07M triethanolamine = pH 2.5 can be more appropriate.3
Mobility matching of the peaks and buffer components is very important in indirect UV detection.4 For example, background UV-absorbing species, such as chromate, are used to match the migration speed of sulphate and chloride ions whereas species such as imidazole are used to mobility match with metal ions such as sodium and potassium.
What About the Buffer Strength?
Higher ionic strength buffers can improve peak shape as a result of "sample stacking" and capillary wall shielding. However, higher ionic strengths also increase the current generated. Methods are, therefore, generally optimized using a compromise of buffer strength, temperature, applied voltage and capillary dimensions.
"It is recommended that the operating conditions, including electrolyte concentration, are adjusted to maintain current levels below 100 μA — above this self heating occurs within the capillary and methods become highly unstable."
Web Information
There is extensive information on buffers and details on the website www.sigmaaldrich.com/. Specifically at http://www.sigmaaldrich.com/Area_of_Interest/Biochemicals/Buffer_Explorer.html there are buffer charts and tools to calculate buffer recipes.

Figure 2
There is also a very useful buffer recipe application at http://www.liv.ac.uk/buffers/buffercalc.html Figures 2–3 shows screenshots of sites that help to specify exact buffer recipes.

Figure 3
Commercially Available Buffers?
CE instrument suppliers and consumable suppliers (www.ceandcec.com/suppliers.htm) sell standard buffers such as phosphate and borate and specialist buffers for kits and specific applications. Figure 4 shows the range of buffers available from Beckman. Sigma-Aldrich and other fine chemical suppliers also sell pre-made buffer solutions.


Figure 4
Common Problems in Buffer Preparations
Dilution of stock pH adjusted buffer: A common laboratory practice in buffer preparation is to prepare concentrated buffer solutions. These stock solutions are then diluted to obtain the required run buffer concentrations. This experimental problem was examined by initially preparing a stock solution of 2 M sodium borate adjusted to pH 9.4.2 This solution was then diluted with water to obtain a final concentration of 500 mM sodium borate. The pH was re-examined and was found to be 9.33.
The same procedure was repeated on a 1 M solution of sodium di-hydrogen orthophosphate adjusted to pH 2.50 with concentrated phosphoric acid (~15 M). This solution was diluted with water to obtain a final concentration of 500 mM and the pH was 2.58.
"Good working practice would be the preparation of a buffer at the required pH rather than diluting a stock solution."
Buffer description errors: Another common problem is the description of preparation procedures given in the literature, where many research papers in CE include a simple but vague description of the buffer composition. Consequently, an analyst attempting to repeat the work is regularly faced with an inadequate description. Taking the term "borate" as an example, this description is extremely ambiguous and has appeared consistently in the literature. For example, sodium borate solution (15 mM) was used in the separation represented in Figure 4(a) and sodium tetraborate (15 mM) in Figure 4(b) for resolution of the acidic test mix.2 Because 1 mole of tetraborate is equivalent to 4 moles of borate then 15 mM tetraborate will be equivalent to 60 mM borate.
The migration times are very different as the tetraborate solution is four times more concentrated in borate ions, thus generates a larger current and longer migration times.
"It is strongly suggested that buffer solution preparations are described in exquisite detail to ensure consistent preparation."
Problems during pH adjusting procedure: A frequent problem occuring in buffer preparation is ''overshooting'' the required pH, which will also alter the ionic strength of the buffer. For example, for a phosphate buffer of pH 7.0 that is adjusted with concentrated phosphoric acid, an excessive amount of acid can be added. This then requires the addition of base to bring the pH back to the required value. This buffer will then have a different ionic strength to that of a buffer prepared exactly to the required pH. This was confirmed by correctly preparing a solution of di-sodium hydrogen orthophosphate to pH 7.5 with concentrated phosphoric acid (15 M).2 A second solution of di-sodium hydrogen orthophosphate was deliberately adjusted to pH 6.0 and then readjusted back to pH 7.5 with 1M sodium hydroxide (NaOH). Both buffer solutions were then used to analyse a test mixture containing methimazole, warfarin, prenidisolone and naphthoxy acetic acid at 100 μg/mL disolved in water (n = 10). The correctly prepared buffer provided a lower current (68 μA) and more precise (run-to-run) migration times than the incorrectly prepared buffer, which generated a current of 75 μA (Table 4).

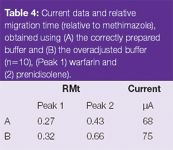
Table 4: Current data and relative migration time (relative to methimazole), obtained using (A) the correctly prepared buffer and (B) the overadjusted buffer (n=10), (Peak 1) warfarin and (2) prenidisolene).
Not specifying when to measure pH: Addition of organic solvents to buffers alters the number of protons in solution and, therefore, invalidates the true measurement of pH. Consequently, it is advisable to specify measuring the pH before the addition of a specific volume of solvent. A full method should specify when to measure pH — a good recent example specifies that the pH should be measured before adding cyclodextrin to the buffer5 — sulphated cyclodextrins are acidic and will lower the pH when added. Heat is usually generated when acids and bases are mixed. Buffer solutions should be allowed to reach room temperature (after mixing or removing from a fridge) before measuring pH as pH is temperature dependent.
Incorrect use/care of pH meter: Manufacturers of modern pH meters have made operation so simple that successful operation is often taken for granted, but it is not quite as easy as it looks. Electrodes must be clean and properly filled. Calibration buffers must be fresh and span the pH range of interest. Temperature is an important factor; not only does the pH of most buffered systems change with temperature, the response of a pH electrode has a temperature dependence of its own. The temperature setting on a pH meter is used to adjust the temperature of the electrode during the measurement, not to correct the temperature where the buffer solution will be used. Specific volumes of specific concentrations of acids and bases can be mixed to make pH values, for example, phosphoric acid and triethanolamine, which avoids use of a pH meter.3
"The best way to use a pH meter is to avoid its use — it is more reliable to mix measured volumes of acids and bases together to generate a specific pH rather than rely on a pH meter."
Buffer not used within its buffering range: A buffer is only a buffer within 1 pH unit of its pKa value; working within 0.5 pH units of pKa is better for critical work. A buffer is needed not only to provide an appropriate pH for the desired process but also to maintain that pH in the face of outside influences such as temperature changes, reaction product formation or factors entering from the environment (i.e., gases such as CO2).
However, it has been found during literature investigations that many methods cited6,7 do not use the buffer within their adequate range, for example, phosphate pH5 is an electrolyte solution not a buffer.6 As a result, in some instances, low capacity buffers used require frequent buffer replenishment because they are quickly affected by "buffer depletion".
Inconsistent reagent grades or descriptions: The grade and source of reagents should be specified because this affects the purity (and cost) of the reagents such as cyclodextrins and sodium dodecyl sulphate. Variations in purity will lead to alterations in ionic strength, current, separation selectivity and, hence, lack of method robustness.
Not specifying shelf-life and storage conditions of buffers: Shelf-life for buffers cannot be assumed. They must be experimentally assessed and proven before writing into methods. For example, materials such as pthalates and metal ions can leach from containers, microbial growth can occur in buffers containing carbohydrate sources such as cyclodextrins or carbon dioxide can be absorbed from the atmosphere.
Insufficiently described buffer filtration procedure: Generally all electrolytes (and sample) should be filtered through at least a 0.45 μm filter to remove particulates, which would appear as apparent noise on the detector baseline. The buffer filtration procedures can result in removal, or addition, of components to the buffer. This may be a result of adsorption onto the filter or extraction from the filter. The filtration process should be assessed, validated and stated in the method. Failure to do this can result in non-robustness. For example, it was observed that up to 15% of the sodium dodecyl sulphate (SDS) was being retained using certain filter types when filtering small volumes.8
"The exact filtration process should be described in the method including the volumes filtered and the filter type used."
"CE Currents" editor Kevin D. Altria is senior principal scientist at GlaxoSmithKline, Harlow, Essex, UK, and is a member of the Editorial Advisory Board of LCGC Asia Pacific.
Direct correspondence about this column to "CE Currents", LCGC Asia Pacific, Advanstar House, Park West, Sealand Road, Chester, UK, CH1 4RN, e-mail: dhills@advanstar.com
References
1. R.J. Boughtflower et al., Chromatographia, 40, 329 (1995).
2. M.A. Kelly unpublished data.
3. Sanger-van de Griend et al., J.Pharm. Biomed. Analysis, 41, 77–83 (2006).
4. M. Macka et al., LCGC N. Am., 19(2), 178–188 (2001).
5. M. Hammitzsch, R.N. Rao and G.K.E. Scriba, Electrophoresis, 27, 4334–4344 (2006).
6. B.J. Clark and A. Shafaati, Anal. Proceed., 30, 481 (1993).
7. C. Schwer and E. Kenndler, Anal. Chem., 63, 1801 (1991).
8. M.A. Kelly, K.D Altria and B.J. Clark, J. Chromatogr., 781, 67–71 (1997).

























Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.

