Application of Quality by Design Principles to a Pharmaceutical Sample Using UHPLC Method Development with Modeling Technologies
Special Issues
A new type of method development that uses modeling to find the "best" separation for high performance liquid chromatography (HPLC) was investigated and principles of Quality by Design (QbD) were followed when planning the investigation. The process delivered precise results and the method was able be transferred to a routine quality control (QC) laboratory.
A new type of method development that uses modeling to find the "best" separation for high performance liquid chromatography (HPLC) was investigated. Principles of Quality by Design (QbD) were followed when planning the investigation. A simple Design of Experiments (DoE) with only three measured factors — gradient time, pH, and temperature — was used with different columns. The basic experiments were saved in an electronic file with the peak tracking results. After calculating a Design Space, the best prediction was compared with a confirmation run. The process delivered precise results and the method was able be transferred to a routine quality control (QC) laboratory.
For many years, a trial-and-error approach has been commonly practiced in high performance liquid chromatography (HPLC) method development — a "pick the winner" strategy. After the validation process had begun, several surprising observations were often made such as new peaks, the disappearance of other peaks, and changes in critical peak pairs. The typical response was to go back to the development process. To try to improve the separation, several test steps would be carried out, to interject quality into the method. This time-consuming process can be avoided by applying Quality by Design (QbD) principles, ensuring the quality of the method at the design stages (1). A well-established means of implementing QbD principles is to use modeling software.
To apply QbD principles to the development of HPLC methods, focus must be placed on method understanding and solid science. We took two-dimensional modeling and extended it to include a third measured factor, establishing the "Cube" (2). In a 2011 paper we demonstrated that predictions made in this way are of high quality (3). In QbD-related method development processes, trial-and-error approaches should be avoided. The best method should be developed in a planned manner, based on solid science (4). There is a great number of different factor combinations that influence retention and resolution of the components in a mixture. The first step is column selection (5). In this study, we tested over ten different columns and selected the Acquity BEH C18 column (Waters). To establish the number of peaks, the sample was first tested with a "scouting gradient."
Which Design of Experiments (DoE) is the simplest? The most successful design is the tG-T model (where tG = gradient time and T = temperature). The most successful design is simple because it requires only four runs: two gradients, tG1 and tG2, with a difference of three in tG (or the slope), as was shown by Snyder and Dolan (6). We extended this design with the addition of a third factor, either pH or ternary composition, tC (2). The columns used had dimensions of 50 mm × 2.1 mm packed with 1.7-μm particles. We also used 4 and 12 min run times, with two different temperatures, T1 and T2, here 20 °C and 50 °C. The four runs were performed following a simple batch protocol. The main points are: not to change the injection volume during the investigation; not to interrupt the process if new peaks appear; and to use a representative sample containing all possible, even unknown, components.
The next step was to organize the received chromatograms and make sure peak movements were realized and understood — a process called peak tracking. Every component is aligned in a horizontal line. This can be supported by the peak area of the sample, which is fairly constant in a tG-T model. In the case of overlapping peaks, the areas are additive.
When the peak table was finished, the software calculated mathematical functions for each component. It allowed the visual examination of the resulting chromatograms, including peak movements, peak overlaps, and changes in elution order.
Compound retention behavior is modeled so that the influence of all significant parameters (alone and in combination) is known, quantified, and visualized (2). In the HPLC method development process, relationships between parameters and their effect on the results are in this way well understood. This translates into a richer chromatographic understanding and facilitates a better scientific work.
Rather than running random test arrangements — necessary when applying statistical analysis — to fulfill QbD conditions, HPLC method development with three-dimensional models is achieved by running three tG-T models with only 12 simple experiments. From these, a large number (over 106) of virtual experiments could be derived and the best one found quickly. The obtained retention models were used for robustness testing, continuous quality control, and method transfer, and could serve as a fundament for Knowledge Management. In the following, we planned to compare an older method that took approximately 40 min with the new approach, based on a practical example from an industrial production process, using tG-T-pH studies to enhance column selectivity choices in ultrahigh–pressure liquid chromatography (UHPLC).
Experimental
Eluent
The mobile phase was a mixture of acetonitrile and 5 mM ammonium dihydrogen phosphate buffer. Acetonitrile (gradient-grade), methanol, ammonium dihydrogen phosphate, phosphoric acid, and standard reference buffers (pH 2.00, 4.01 and 7.00) were obtained from Merck. For measurements, water was prepared fresh using ELGA Purelab UHQ water. The buffer was filtered before use on a regenerated cellulose filter membrane, 0.2-μm pore size (Sartorius).
Samples contained 10–30 μg/mL amlodipine and its European Pharmacopeia (Ph.Eur.) impurities (A, B, D, E, F, G, H) and 1 mg/mL amlodipine spiked at 0.1% (European Directorate for the Quality of Medicines & HealthCare).
Equipment and Software
An Acquity UHPLC system (Waters), Empower software, and Acquity BEH C18 50 mm × 2.1 mm, 1.7-μm column (Waters) were used. This system had a 5-μL injection loop and a 500-nL flow cell. The dwell volume of the system was measured to be 0.125 mL. The pH was measured with a MP 225 pH meter (Mettler Toledo).
Method development and method modeling were performed using DryLab 2010 HPLC/GC optimization software, consisting of the DryLab Core-module v.3.9, PeakMatch v.3.6, and the 3D-Resolution Space feature called the "Cube" (Molnár-Institute).
Results and Discussion
The problem of older HPLC methods, which may be available in pharmacopeias, is that the columns are often of an old type which are sometimes not available anymore. Furthermore, many old methods are very slow. This causes, in a modern pharmaceutical environment, great delays in the production of certain drugs. We need an easy replacement for older methods to be able to use more modern instrumentation and columns. We tried, in our case, to reduce the analysis time of the old method from 40 min to hopefully less than 10 min.
There were several approaches to the selection of the proper design in the literature (7,8). As DoE we use the one shown in Figure 1 with 12 experiments (2). The advantage here is the fast evaluation of the sample using gradients. The creation of a cube was based on reversed-phase gradient elution (9) with the columns mentioned above, and was done in 3-4 h, representing more than 106 virtual experiments. Column selectivity was evaluated quickly under consideration of both stationary and mobile phase influences.


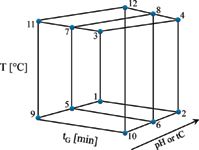
Figure 1: Design of experiments (DoE) for the simultaneous optimization of gradient time (tG), temperature (T), and pH of the eluent (A); or ternary composition (tC) of the eluent (B). Circles represent the twelve experiments inputted for the 3-D model.
Experimental design for simultaneous optimization of gradient time, temperature, and pH and ternary eluent composition tC, required 12 experiments, as illustrated in Figure 2 (2). Two basic gradients with different rates (4 and 12 min gradient time, in a range of 30 – 90% B) were carried out at two different column temperatures (20 and 50 °C). The mobile phase "A" consisted of 5 mM ammonium dihydrogen phosphate buffer (pH = 2.0, 2.5 and 3.0), "B1" was acetonitrile, and "B2" was methanol. The flow-rate was at 0.5 mL/min. The injection volume was 1 μl.


Figure 2: Experimental designs for the simultaneous optimization of gradient time, temperature, and pH for three organic solvent compositions. Circles represent 12 experiments input for the 3D model.
We were able to create a four-dimensional model, as we prepared three cubes in identical pH ranges, but with three different ternary eluents, methanol, acetonitrile and 50:50 (v/v) (methanol:acetonitrile). Then we selected a pH value, where our separation was robust in terms of pH with little risk of losing efficiency. From the three cubes we exported 3 × 4 chromatograms as the reference points of a new cube.
Predicted optimum parameters were: tG = 5 min; gradient range: 30–70% B; T = 40 °C; pH = 2.3; and ternary composition (tC)= 75:25 (v/v) acetonitrile:methanol.
From three cubes we selected those tG-T sheets that belong to pH 2.3, as shown in Figure 3.


Figure 3: Three-dimensional tG-T-pH models (Cubes) with three different organic solvents. Red colors mean regions above Rs (crit > 3.0), blue colors indicate co-elution (Rs,crit = 0) of the closest ("critical") peak pair.
In Figure 4 we can see that the system is robust for pH changes, but not robust at two ternary eluents: With 100% acetonitrile (AN) the resolution between ImpG and ImpB is small. In the case of methanol (MeOH) the components ImpE and ImpG exhibit partial overlaps. Using 50:50 (v/v) methanol:acetonotrile as eluent B, we achieve a much better separation of the critical impurity components.


Figure 4: Predicted chromatograms are showing that in all three eluent compositions robust separations are possible at pH 2.3 (tG = 8 min, 30%â70% B, T = 40 °C).
The next step was the selection of the "working point" at 40 °C and tG: 5 min (30–90% B) where B was 75:25 (v/v) acetonitrile:methanol.


Figure 5: Four-dimensional tG-T-ternary model (fourth dimension was pH) and tG-T-sheet with "working point".
Finally, the conformation of the prediction in Figure 6a revealed an excellent separation of all components, which could be well confirmed, as shown in practice with a mix of the standards in Figure 6b and with the drug product in Figure 6c.


Figure 6: (a) Predicted chromatogram, (b) experimental chromatogram, (c) experimental spiked chromatogram at 0.1% level.
The precision of the predicted vs. modeled experiments are showing only small deviation of a few hundredths of a min (11–13). Looking at the last two peaks, the retention times are 3.145 vs. 3.148 min, a difference of 0.003 min and 4.944 vs. 4.911, a difference of 0.033 min.


Figure 7: Experimental designs for simultaneous optimization of gradient time, temperature, and ternary composition at three different pH values of the eluent A. Circles represent the 12 experiments input for the 3D model.
After these steps we turned the process in the experimental design around and produced first three Cubes with a constant ternary composition region, but with three different pH values. For the creation of a new Cube we selected one ternary composition, and from the three Cubes we exported 12 runs. We received at the same working point the same separation as in the first part of the study (see Figure 7–10).


Figure 8: Three-dimensional tG-T-ternary models (Cubes) with three different pH values (2.0, 2.5, and 3.0). Red colors mean regions above Rs (crit > 3.0); blue colors indicate coelution (Rs, crit = 0) of the closest ("critical") peak pair.
In Figure 9, the "working point" was selected at tG = 5 min and T = 40 °C and pH 2.3. The predicted chromatogram has a run time of less than 5 min.
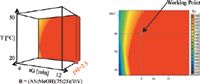
Figure 9: Four-dimensional tG-T-pH model (fourth dimension was ternary composition) and tG-T-sheet with "working point".
Using the software we were also able to increase the speed of the separation by modeling the flow rate.
With a faster gradient time (tG = 2 min) and an increased flow rate of 0.8 mL/min, we were able to further reduce the analysis time to less than 1.8 min.

Figure 10: Predicted four-dimensional chromatogram.
The new predicted parameters are: tG = 2 min; gradient range 30–90% B; flow rate: 0.8 mL/min; T = 40 °C; and pH = 2.3; and with tC = 75:25 (v/v) acetonitrile:methanol we receive the prediction shown in Figure 11a with the corresponding confirmation run in Figure 11b. In this way, the analysis time could be reduced to less than 1.8 min.

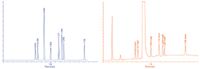
Figure 11: (a) Predicted chromatogram with increased flow rate to 0.8 mL/min; (b) Experimental spiked chromatogram at 0.1% level. Conditions are as described in the text for Figure 11(a).
The Ph.Eur. method is very long and this specifies only three impurities (14) while our method takes less than two minutes and seven impurities are specified (see Figure 12).

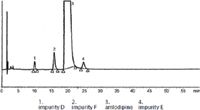
Figure 12: Chromatogram of amlodipine and its impurities from the European Pharmacopeia 7.4, Amlodipine besilate 04/2012:1491 p.4275-4276. The Ph.Eur. method is very long and specifies only three impurities, while our method has a run time of less than 2 minutes and specifies seven impurities.
Summary
In this study, we were able to reduce the analysis time of a pharmaceutical preparation from 40 min to below 1.8 min using UHPLC instrumentation, modern short UHPLC columns, and modeling software. With the short analysis time the production of the drug is faster and more economical than in the past. The use of the modeling software allowed us to fulfill QbD criteria for the method, increasing flexibility in a routine operation. Retention and critical resolution phenomena became more transparent than they were before. Method transfer is also easier using the Knowledge Management protocol described in the paper. In case of renewing older methods we recommend the above technology because of its reliability and speed.
References
(1) International Conference on Harmonization ICH Q8(R2) – Guidance for Industry, Pharmaceutical Development (2009).
(2) I. Molnár, H.J. Rieger, and K.E. Monks, J. Chromatogr. A. 1217, 3193–3200 (2010).
(3) I. Molnár, K.E. Monks, H.J. Rieger, and B.T. Erxleben, LCGC The Column 7(5), 2–8 (2011).
(4) M. Nasr, CDER, FDA, Lecture on "Quality by Design (QbD): Analytical Aspects" at HPLC 2009, Dresden, Germany, Sept 2009.
(5) L.R. Snyder, J.W. Dolan, and P.W. Carr, J.Chromatogr. A. 1060, 77–116 (2004).
(6) J.W. Dolan, L.R. Snyder, T. Blanc, and L.Van Heukelem, J.Chromatogr. A. 897 37–50 (2000).
(7) L.R. Snyder, J.J. Kirkland, and J.W. Dolan, Introduction to Modern Liquid Chromatography (John Wiley and Sons Inc. New Jersey 2010).
(8) A.Y. Kazakevich and R. LoBrutto, HPLC for pharmaceutical scientists (John Wiley and Sons Inc. New Jersey 2007).
(9) C. Horváth, W. Melander, and I. Molnár, J. Chromatogr. 125, 129–156 (1976).
(10) L.R. Snyder and J.L. Glajch, Computer-Assisted Method Development for High-Performance Liquid Chromatography, J.Chromatogr. Library, 485(1989), pp. 1–640.
(11) I. Molnár, J. Chromatogr. A. 965, 175–194 (2002).
(12) I. Molnár and K. Monks, Chromatographia. 73 (2011) (Suppl.1) S5–S14.
(13) M. Euerby, G. Schad, H.J. Rieger, and I. Molnár, Chromatography Today 3(13)(2010)
(14) European Pharmacopeia 7.4, Amlodipine besilate 04/2012:1491, 4275–4276.
Dr. rer. nat. Imre Molnár, president of Molnár-Institute, is a former coworker of Prof. Csaba Horváth at Yale University, where he developed the fundamentals of reversed-phase chromatography and the well-known theory of "solvophobic interactions." In 1981 he founded the Molnár-Institute for Applied Chromatography in Berlin, Germany. He specializes in pharmaceutical research and analysis and conducts a series of HPLC courses on reversed-phase HPLC and the development of robust HPLC methods. Since 1984 he has worked with LC Resources Inc. on the development of the DryLab software, which is now widely used in industry and in the life science community as a modeling tool for complex separations, enabling the user to minimize the number of experiments required to develop an optimized and robust method. Since 2006 DryLab has been developed in Berlin further; the new version DryLab 4 is written in C#. Direct correspondence to: imre.molnar@molnar-institute.com
Dr. rer. nat. Hans-Jürgen Rieger, vice president and product manager, has been working with the Molnár-Institute since 1999 as a chemist, specializing in software programming. In cooperation with Dr. Molnár, he is responsible for the development of new modules of DryLab for better peak tracking, visualizing the Design Space, and robustness calculations as well. He also teaches DryLab-courses on an international scale. Hans-jürgen.rieger@molnar-institute.com
Róbert Kormány is a chemist with a Master of Science degree from the University of Debrecen in Hungary. He completed his education with an additional degree in chromatography and analytical chemistry from the Institute of Analytical Chemistry of the Technical University Budapest in the department of Prof. Dr. Jenö Fekete. Mr.Kormány specializes in UHPLC method developments for the separation of pharmaceutical compounds using computer simulation. E-mail: rkormany@gmail.com






















Separating Impurities from Oligonucleotides Using Supercritical Fluid Chromatography
February 21st 2025Supercritical fluid chromatography (SFC) has been optimized for the analysis of 5-, 10-, 15-, and 18-mer oligonucleotides (ONs) and evaluated for its effectiveness in separating impurities from ONs.

