Eliminating Bias in the Resolution and Analysis Time During the Simultaneous Determination of Anionic and Cationic Species in Capillary Zone Electrophoresis
Special Issues
A demonstration of how to successfully convert from capillary zone electrophoresis to dual-opposite injection capillary zone electrophoresis for routine experiments is outlined.
A simple approach to eliminate bias in the resolution and analysis time during the simultaneous determination of anionic and cationic species in capillary zone electrophoresis (CZE) is proposed using dual-opposite injection capillary zone electrophoresis (DOI–CZE). DOI–CZE is well-suited for the analysis of oppositely charged analytes, especially if there is a difference in the concentration of the analytes. The use of very different migration distances can eliminate co-detection. A method for the simple conversion of CZE to DOI–CZE is presented to demonstrate the ease at which DOI–CZE could replace CZE in routine experiments.
Capillary zone electrophoresis (CZE) is a voltage-driven separation technique that separates charged analytes with high efficiency, low sample consumption, and short analysis time. These advantages over other common separation techniques, such as high-performance liquid chromatography (HPLC) and gas chromatography (GC), have made CZE an attractive method to separate a wide range of complex mixtures.
In conventional CZE experiments, fused-silica capillaries with inner diameters of 25–75 μm and lengths of 30–100 cm are commonly used. Separation is achieved because of differences in the charge and size of the analytes of interest. Bulk flow is provided by electro-osmosis pushing all analytes, regardless of charge, toward a downstream detector. When a voltage is applied, electro-osmotic flow (EOF) is cathodic (that is from anode to cathode).
As a result, cations travel through the capillary in the same direction as the EOF toward a detector located near the cathodic end of the capillary. The neutral analytes are pushed through the capillary with the same velocity as the EOF, but are not separated from one another. Anions, attracted to the anode, migrate in the opposite direction of the EOF. For anions to be detected, the EOF must be of high enough magnitude to push the anions toward the detector.


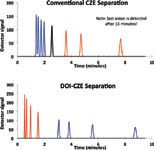
Figure 1: Simulated electropherograms of (a) a CZE and (b) DOI-CZE separation of a mixture of four cations (blue), four anions (red), and one neutral analyte (black). Parameters used to generate these electropherograms are shown in Figure 2. Note that only three out of four anions are eluted within 10 min in the CZE mode.
A simulated CZE electropherogram, shown in Figure 1a, was generated based on values shown in Figure 2 to emphasize problems commonly encountered in CZE experiments. Figure 1a shows that cations migrate through the capillary much more quickly than anions and, therefore, have less time to be resolved into their individual peaks than anions with slower migration.


Figure 2: Conditions used to generate the simulated electropherograms shown in Figure 1.
Some anions can exhibit extremely slow migration times and for the smallest, fastest anions, may not reach the detector at all. For example, Anion 4, with a migration time of 15.33 min, cannot be displayed in Figure 1a. If EOF is reversed (from cathode to anode), anions elute first with poor resolution and cations elute slowly or not at all (1). These observations provide evidence of a strong bias in the migration time and resolution of anions and cations, when simultaneously separated in a single run.
To eliminate bias, most researchers simply separate cationic or anionic analytes, and optimize either resolution or analysis time. The separation usually involves moderate to high EOF and electrophoretic migration in the same direction as the EOF. However, this practice is rather disadvantageous because two separate analyses and optimizations are required to analyze a mixture containing both anions and cations. It would be much more time and resource efficient to do the separation in one experiment.
Dual-opposite injection capillary zone electrophoresis (DOI–CZE) is a less commonly known form of capillary electrophoresis. It was first reported in 1998 by Kuban and Karlberg and independently by Padarauskas and colleagues for the separation of small inorganic anionic and cationic analytes (2,3). The technique has since been expanded to include the separation of organic species and common pharmaceutical compounds. In DOI–CZE, sample is injected from both ends of the capillary under conditions of suppressed electro-osmotic flow. The combination of dual-opposite end injection and suppressed EOF allow anions and cations to migrate from opposite ends of the capillary toward the electrode of opposite charge. When UV absorbance detection is used, the detector window is typically located near the center of the capillary. The position of the detector window can be optimized for best resolution, fastest analysis time, and/or avoidance of co-detection.
Figure 1b depicts a simulated electropherogram of the same analytes as Figure 1a that has been generated using DOI–CZE experimental conditions. Under DOI–CZE conditions, the anions elute first because they travel the shorter distance to the detector window. It is important to note that unlike CZE, the first anion to elute under DOI–CZE conditions is the one with the fastest electrophoretic mobility, as a result of the low EOF. In CZE, the anion with the fastest velocity elutes last because it is more successful in migrating against the high EOF. In the simulated electrophoretic data for CZE (Figure 1a), the fastest anion is not detected until after 15 min. Neutral analytes are generally undetected in DOI–CZE because there is little to no EOF to push them toward the detector. The fastest cation is detected shortly after the slowest anion in an effort to provide the fastest possible separation. If the EOF direction is reversed, the elution order would change because the effective capillary lengths would also change. As can be seen by these simulations and elsewhere (4), there is no observed bias in the migration time and resolution between anions and cations when simultaneously separated in a single run under DOI–CZE conditions.
In DOI–CZE, both the analyte electrophoretic mobilities and the capillary lengths on either side of the detection window can vary widely. Therefore, it becomes possible that an analyte migrating from one end of the capillary can be detected at exactly the same time as an analyte migrating from the opposite end of the capillary. When co-detection (resolution of zero) occurs, several approaches can be used to avoid future problems (4). First, a new capillary can be made with a repositioned detection window. However, this can be both costly and time consuming. Second, a small amount of external pressure (for example, 5 mbar) can be applied to either end of the capillary during the analysis to generate just enough forward or reverse hydrodynamic flow to change the migration times of the previously co-detected ions without introducing a measurable amount of zone broadening. Third, a delay can be incorporated into the injection sequence during which a preliminary voltage is applied, allowing analytes of one charge to migrate toward the detector for a specified amount of time before the second sample is injected and the separation voltage is applied. However, optimizing the conditions for any of these approaches to avoid co-detection of one combination of oppositely-charged analytes may result in co-detection problems for another combination, especially when the number of analytes is large relative to the peak capacity.
A previously unreported fourth approach is to use a capillary with very different migration distances to the detector window from the two ends of the capillary. This ensures that all analytes of one charge have passed through the detection window before any analytes of the opposite charge. The resolving power for the analytes traversing the shorter migration distance will be lower, but is more than compensated for by the universal elimination of co-detection.
To simplify the determination of appropriate migration distances, we have derived relationships to calculate the effective lengths needed to avoid co-detection of the fastest cation and slowest anion (or vice-versa). First, k, the ratio of effective lengths that would actually result in co-detection, is calculated,

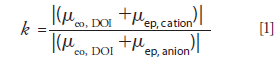
where μeo, DOI is the electro-osmotic mobility under DOI–CZE conditions, μep, cation is the largest cationic electrophoretic mobility, and μep, anion is the smallest anionic electrophoretic mobility. Alternatively, the length ratio could be based on the smallest cationic electrophoretic mobility and the largest anionic electrophoretic mobility. All of the mobilities can be measured experimentally.
Once k is known, the shorter effective length on one side of the capillary, L2 can be chosen based on the design of the CE instrument and L1, the longer effective length on the opposite side of the capillary, can be calculated using the equation


Once the lengths calculated using equations 1 and 2 that would result in co-detection of the fastest cation and slowest anion or vice-versa are known, then either of two possible adjustments can be made to eliminate the co-detection.
The first adjustment would be to modify sample introduction conditions. Detailed information on the three types of sample introduction is given in a paper by Weekley and Foley (4). Briefly, simultaneous electrokinetic injection (SimEI) uses an applied voltage for an allotted amount of time to both ends of the capillary, in which two identical sample vials are placed. After the sample is injected, the capillary is placed into buffer vials and the separation voltage is applied. The two other modes of injection use a sequential injection. In sequential electrokinetic (SeqEI) or sequential hydrodynamic injection (SeqHI), aliquots of the same sample are introduced sequentially at one end of the capillary and then the other. When using sequential sample introduction, the first aliquot of the sample plug could be expelled from the capillary during the introduction of the second aliquot at the opposite end. This can be avoided by moving the first aliquot further into the capillary using a preliminary transport (PT) step before the introduction of the second aliquot. "Preliminary transport" is accomplished by adding a plug of buffer into the capillary behind the first aliquot via a voltage, pressure or both for an appropriate length of time.
Therefore, it is suggested that the first aliquot should be introduced into the end of the capillary with the shorter migration distance to the detector window. If the plug of buffer injected behind the first aliquot is large enough, when the second aliquot is introduced into the other end of the capillary, the first aliquot will be pushed backward slightly but will be ahead of its original position after it was introduced. This therefore modifies the initial effective capillary length to a shorter value. The distance that this first aliquot is pushed into the capillary during the sample introduction sequence can be easily calculated. When SimEI is desired, the options are to either (i) to increase the length of the longer section of the capillary by a small amount (for instance, 1 cm); or (ii) to decrease the length of the shorter section of the capillary by a similar amount.
Equations 1 and 2 make avoidance of co-detection in DOI–CZE relatively easy. Therefore, it is relatively simple to take advantage of the benefits of DOI–CZE by converting a CZE method.
First, to determine if extreme bias exists under the chosen experimental conditions, a CZE experiment must be run. From this, the electrophoretic mobilities of analytes are calculated. Next, a new capillary is made for the DOI–CZE method and the desired method of EOF suppression is selected (coated capillary, buffer additives, low pH). We propose starting with the shortest possible total length capillary for a given instrument to allow the shortest separation time possible. Next, the electro-osmotic flow under suppressed conditions is measured using the method described by Williams and Vigh (5). From the experimentally determined electro-osmotic and electrophoretic mobilities, the ratio of migration distances, k, can be calculated to determine the minimum capillary length needed to avoid co-detection between oppositely-charged analytes migrating under DOI–CZE conditions. Once these values are known, it may be necessary to remake a capillary in DOI–CZE. Finally, the experiments are run under the desired DOI–CZE conditions. If analyte resolution following elution from the short end of the capillary is problematic, both L1 and L2 should be multiplied by the same factor to lengthen the capillary. The appropriate ratio of migration distances between sides should be maintained.
Another benefit of DOI–CZE is the ability to vary the amount of anions and cations injected individually. This method was applied to a real-life sample, Cold & Sinus Liqui-Gels (Advil). One Liqui-Gel capsule contains 200 mg ibuprofen (an anion) and 30 mg pseudoephedrine (a cation). There is almost seven times more ibuprofen than pseudoephedrine in each Liqui-Gel capsule, making it very difficult to detect both analytes in one run in a CZE experiment. If ibuprofen is diluted to an appropriate concentration for detection, pseudoephedrine is extremely difficult to detect because it is present in a lower quantity. If pseudoephedrine is prepared at a detectable concentration, ibuprofen is often too concentrated and elutes as a very broad peak. To solve this problem, we used DOI–CZE with sequential sample introduction to modify the quantity of each analyte injected.
Experimental
All experiments were performed on an HP3D CE instrument, model G1600AX (Agilent Technologies), equipped with a diode array detector. The primary wavelength monitored was 215 nm. ChemStation software (Agilent Technologies), version B.03.02, was used for data acquisition (at 10 Hz) and analysis. Fused silica capillaries (50-μm i.d., 365-μm o.d., 32 cm total length, 23.6 cm effective length, Polymicro Technologies) were used for the CZE experiments and Guarant-coated capillaries (50 µm i.d., 23.6 cm (8.4 cm) from anode (cathode) to detector, Alcor BioSeparations LLC) were used for the DOI–CZE experiments. The window for UV detection in the coated capillary was burned according to the procedure from the manufacturer to avoid destroying the inner coating. The cartridge temperature was maintained at 25 °C for all experiments.
Distilled deionized water was obtained from a Barnstead E-Pure Water System with a resistance of at least 18 MΩ. Sodium phosphate monobasic monohydrate and sodium phosphate dibasic heptahydrate were purchased from J.T. Baker. Ibuprofen was purchased from Sigma-Aldrich and pseudoephedrine was purchased from Spectrum Chemical. Advil Cold & Sinus Liqui-Gels were purchased from a local pharmacy.
The aqueous buffer solutions were prepared by dissolving the appropriate amount of sodium phosphate monobasic dihydrate and sodium phosphate dibasic heptahydrate in distilled deionized water equivalent to approximately three-quarters of the final volume. The total phosphate concentration was 25 mM. Sodium hydroxide was added until the pH reached 7. Water was then added to bring the solution to volume. The same buffer solution was used for both CZE and DOI–CZE experiments.
One Liqui-Gel was placed in 50 mL of buffer overnight to dissolve the outer coating to allow access to the solubilized ibuprofen. Then 50 mL of methanol was added to dissolve the ibuprofen. The final concentration was 2 mg/mL ibuprofen and 0.30 mg/mL pseudoephedrine, which could then be diluted as desired. Ibuprofen and pseudoephedrine standards were made to verify which component from the mixture corresponds to which analyte. Concentrations were kept similar to the final dilution of the Liqui-Gel.
The conditioning of new capillaries for CZE was as follows: distilled deionized water for 10 min, 1 M NaOH for 10 min, 0.1 M NaOH for 5 min, distilled deionized water for 3 min and 25 mM phosphate buffer solution (pH 7) for 15 min. This conditioning was also run at the start of each day. CZE injections were performed hydrodynamically at +15 mbar for 2 s. New capillaries for DOI–CZE were conditioned with only 25 mM phosphate buffer solution (pH 7) for 30 min as recommended by the manufacturer. This conditioning was also run at the start of each day. Both CZE and DOI–CZE capillaries were flushed with buffer for 2 min between runs.
Injections in DOI–CZE were performed via sequential hydrodynamic injection (SeqHI). Most injections consisted of an initial injection of sample at the cathode of –5 mbar for 2 s, followed by an injection of buffer behind the sample at –15 mbar for 5 s. Sample was then introduced to the opposite side of the capillary at +15 mbar for 5 s.
Following the completion of sample introduction and preliminary transport where applicable (DOI–CZE only), a constant applied voltage of 14 kV was used for all experiments after a 0.3 min voltage ramp from 0 to 14 kV. All data were evaluated with a minimum of five successive injections for each data set.
Results and Discussion
The experiment was first run under CZE conditions and the resulting electropherogram is shown in Figure 3. In this experiment, approximately 0.03 mg of pseudoephedrine and 0.21 mg of ibuprofen were injected into the capillary. As expected, pseudoephedrine eluted first in about 1.10 min, followed by ibuprofen detected after 2.36 min. There is limited bias with respect to the migration time and resolution between analytes of opposite charge in this electropherogram as a result of the small number of components analyzed. However, DOI–CZE can be used to increase the amount of pseudoephedrine for better detection.

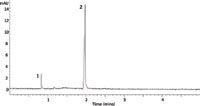
Figure 3: Electropherogram of Cold & Sinus Liqui-Gels (Advil) using capillary zone electrophoresis. Peaks were identified as pseudoephedrine (1) and ibuprofen (2). Conditions are described in the experimental section.
The same sample was then run under the DOI–CZE conditions described in the experimental section. The resulting electropherogram is shown in Figure 4. Based on the injection procedure described above, approximately 0.11 mg of pseudoephedrine and 0.14 mg of ibuprofen were injected. It is less obvious that the peaks are similar in terms of peak heights and/or peak areas as broadened peaks were noted for both analytes (most likely as a result of interaction with the wall of the coated capillary).

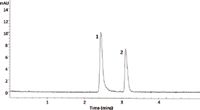
Figure 4: Electropherogram of Cold & Sinus Liqui-Gels (Advil) using dual-opposite injection capillary zone electrophoresis. Peaks were identified as ibuprofen (1) and pseudoephedrine (2). Conditions are described in the experimental section.
The response factor (RF) of each analyte in each technique was calculated,


where Acorr is the corrected peak area (= Auncorrected ÷ gration time) and qinj is the amount of analyte injected. RF values for pseudoephedrine were determined to be 1.68 and 1.77 for CZE and DOI–CZE, respectively. For ibuprofen, values of 2.33 and 2.92 for CZE and DOI–CZE were reported. It is possible that the increase in RF values observed under DOI–CZE conditions are a result of variations in the volume injected. Although injection volume reproducibility was not measured, it is likely that under the more complicated injection sequence utilized under DOI–CZE conditions, the volume injected may vary slightly run to run. Use of an internal standard would likely improve RF values between techniques.
Conclusions
DOI–CZE is an effective approach for the elimination of the unavoidable and significant biases in the resolution and analysis time of anionic and cationic species in CZE when oppositely-charged analytes are determined simultaneously (in a single run). As well as being generally better-suited for the simultaneous analysis of oppositely charged analytes than CZE, DOI–CZE is particularly beneficial when one class of charged analytes is much more dilute than the other. The simple conversion from CZE to DOI–CZE conditions reported here demonstrates the ease at which DOI–CZE could potentially replace CZE in many routine experiments. Successful investigation of further real-life samples will likely propel DOI–CZE into a more widely used separation technique. Given that a significant source of imprecision in CZE is the variation of EOF, DOI–CZE is potentially more reproducible from a migration time perspective since it is performed under conditions where the EOF is suppressed. We are currently investigating this aspect of DOI–CZE along with the simultaneous separation of enantiomeric anions and cations, such as illicit drugs.
References
(1) J.W. Jorgenson and K.D. Lukacs, Anal.Chem. 53, 1298–1302 (1981).
(2) P. Kuban and B. Karlberg, Anal. Chem. 70, 360–365 (1998).
(3) A. Padarauskas, V. Olsauskaite, and G. Schwedt, Journal of Chromatogr. A. 800, 369–375 (1998).
(4) B.S. Weekley and J.P. Foley, Electrophoresis 28, 697–711 (2007).
(5) B.A. Williams and G. Vigh, Anal. Chem. 68, 1174–11780 (1996).
Donna M. Blackney is a PhD candidate in the Department of Chemistry at Drexel University in Philadelphia, Pennsylvania.
Joe P. Foley is Professor of Chemistry and Interim Associate Department Head at Drexel University. Direct correspondence to: jpf26@drexel.edu













: An Interview With Fabrice Gritti")







University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.


Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.

