Landmarks in the Evolution of Ion Chromatography
From the invention of eluent suppression to today's "just add water" concept, pivotal developments over the last 40 years are chronologically highlighted from a chemical and instrumental viewpoint.
A personal perspective on the milestones in the development of ion chromatography. From the invention of eluent suppression to today's "just add water" concept, pivotal developments over the last 40 years are chronologically highlighted from a chemical and instrumental viewpoint. Suggested key references on applications and detailed developments are provided.
In the late 1950s, a few of us at the Dow Chemical Company began thinking about a new form of chromatography that would challenge the old methods of inorganic ion analysis. Those ideas and subsequent research were the precursors of what became known as ion chromatography (IC). I have been privileged to be closely linked to those early endeavors and to be at least an interested observer of the many other important events that followed. From this long perspective I will attempt to identify ideas, inventions, and innovations that have altered the pace of development and the trajectory of IC in the years since our early imaginings. I call these events landmarks.
In other publications (1,2) I have described in some detail how quite unrelated experiences in my early years at Dow influenced the first IC inventions, so for brevity's sake I will jump directly to the first landmark, in 1971.
1971: The Invention of Eluent Suppression
By 1971, we had decided that what inorganic ion analysis needed was a chromatographic technique that would supplant many of the tedious, time-consuming classical wet-chemical methods. At this time, ion-exchange chromatography had made significant contributions to inorganic analytical chemistry (3) but it was usually as an adjunct to wet-chemical methods; chromatography segregated the analytes of interest from interfering species, fraction collectors made "cuts" of the effluent from the ion-exchange column, and the cuts were analyzed by the classical methods. Because the analytes often had widely diverse chemistries and required special analytical techniques, the task of analysis was handled by specialists. It was a slow business and days could go by between the separation and the results. We envisioned a method that would couple fast separation with prompt detection and measurement of analytes by a monitor placed at the column outlet. Desirably, this detector should be "universal" — that is, capable of quantifying ions of widely diverse chemistries. But what might we use as a detector? Many inorganic ions of interest such as alkali and alkaline earth metal ions, ammonium, halides, nitrate, sulfate, and phosphate were notably "bland" in not having useful chromophores or a general postseparation means of generating chromophores, as Moore and Stein had done for amino acid analysis (4), so spectrophotometric methods of detection seemed a no-go at that time. We recognized, however, that a universal property shared by aqueous electrolyte solutions was electrical conductance and a notable landmark was our decision to exploit that property. But therein we anticipated a problem.
With the considerable knowledge available to us (3), we were convinced that ion-exchange chromatography could separate any mixture that challenged us. We were also aware that using conventional high-capacity ion-exchange resins would in many cases require quite concentrated (several molar) solutions to displace the more intractable species and any conductance changes imparted to the effluent by the appearance of the analyte ions could easily be overwhelmed by the conductance "noise" of the eluent. So we decided — before 1971 — to abandon ion-exchange chromatography and instead develop stationary phases that would separate electrolytes using water as eluent, because water was the perfect background for sensitive conductance measurement. Our early efforts to devise such media were spasmodic and mostly barren of success.
In the fall of 1971, we made a number of significant decisions that led to a breakthrough: We would revert to ion-exchange chromatography as the separation mode and to conductometry as the detection and measurement mode, but instead of placing the conductivity cell right after the separating column, we would insert a second column between the separator and the cell. Initially we called this column the stripper because its purpose was to strip the effluent of the highly conducting eluent and leave just the analytes in water, the ideal background for detection. For example, using the workflow depicted in Figure 1, we would separate the alkali metals on a cation-exchange resin using hydrochloric acid as the eluent, then pass the effluent from the separating column through a bed of anion-exchange resin in the hydroxide form, which would strip out the acid and present the alkali metals to the conductivity cell as the metal hydroxides in a water background. Analogously, anions could be separated on an anion exchanger using sodium hydroxide as eluent, while a following bed of cation-exchange resin in the hydronium form would remove the sodium hydroxide, thus presenting the anions (such as the halides) to the conductivity cell as their acids in a water background.


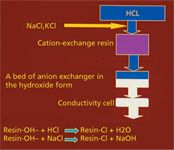
Figure 1: Workflow devised in 1971 using a "stripper" column between the separator column and the conductivity cell, as applied to the separation of the alkali metals on a cation-exchange resin using hydrochloric acid as the eluent.
Thus, we would solve the problem of eluent conductance noise simply by removing the eluent and retaining the analytes in a water background.
As a first implementation of this new idea, we proposed using a strong acid cation-exchange resin (Dowex 50, Dow Water and Process Solutions) as separator and a weak-base resin (Dowex 30) to absorb the HCl eluent. But that idea was never tried — for the following reason.
In using the stripper, we were proposing something radically new for chromatography, a component that would become partially depleted with each sample injected and eventually would have to be regenerated. We felt that for this technique to succeed, this regeneration would have to be as unobtrusive as possible; users should be able to run many samples before stripper regeneration became necessary. It became clear that if we used our first proposal we could easily encounter cases where the elution of even a single sample would exhaust a stripper that at the same time had to be massively larger in volume than the separator to supply enough stripping capacity. The large void volume of the stripper bed would be the source of two serious defects: It would greatly prolong elution times and seriously degrade chromatographic efficiency. Clearly, this was not going to fly.
But the problem suggested a solution (5,6): Use separator columns with very low specific capacity, thus enabling elution with low concentrations of eluent; this in turn would prolong the life of the stripper and thus allow the analyses of many samples before regeneration became necessary. And we envisioned that an effective suppressor could be about the same size as the separator, thus minimizing the problems caused by the suppressor void volume. These concepts were central to our invention of IC with eluent suppression and conductometric detection (7).
Separation Media and Eluents for Early IC
The first successful separation by IC was the separation of a mixture of lithium, sodium, and potassium using a lightly sulfonated styrene–divinylbenzene (DVB) polymer as the separator, dilute hydrochloric acid as eluent, and Dowex 1 (hydroxide form) as the stripper. I had used much more separator than necessary so elution times were unnecessarily long, but otherwise the separation looked excellent, and for that era, the detectability levels were impressive (Figure 2).

Figure 2: The first chromatogram using eluent suppression and conductometric detection. The sample injected was 0.1 mL of a mixture of LiCl, NaCl, and KCl, 0.01 M of each. The eluent was 0.02 M HCl. From author’s laboratory notebook, dated November 9, 1971.
Because of the very basic eluents anticipated for anion analysis, styrene-based anion exchangers with their great chemical stability were preferred over the silica-based media that were being widely used in high performance liquid chromatography (HPLC). However, although surface sulfonation of styrene-based polymers gave a useful cation separator and surface quaternization of a styrene–DVB polymer seemed the obvious route to a low-capacity anion-exchange resin, I saw it as a route fraught with problems. If I used the same synthetic steps as were used to produce high-capacity resins, I anticipated serious obstacles to creating, on a styrene-based substrate, a thin anion-exchanging shell that would not have a diffuse boundary, and diffuse boundaries were anathema to efficient chromatography. Instead, a prior experience had impressed on me the Velcro-like attachment that anion-exchange resins formed with their cation-exchanging counterparts (1,2) so I created the first useful anion separator by treating a surface-sulfonated styrene–DVB resin with a suspension of a colloidal anion exchange resin (Figure 3). Because of the manifold advantages of preparing IC media in this way (1,2), particularly to the manufacturer, stationary phases prepared by this means (8) would become, and for many years remain, the workhorse separating media for anion analysis by IC.

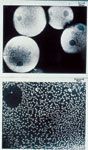
Figure 3: (Bottom) Colloidal anion-exchange particles (about 300 nm in diameter) on surface-sulfonated styreneâDVB particles (about 50 µm in diameter) (top).
Improvements in cation IC came rapidly in our first year but progress in anion analysis was slower. Although sodium hydroxide was an excellent choice in that the stripper action produced the ideal background, the hydroxide ion was an anion of low ion-exchange affinity for anion-exchange resins of that time and high concentrations were required to displace many analyte ions. This high concentration of sodium hydroxide placed a heavy burden on the stripper. This burden was greatly alleviated by using the phenate ion as the eluting anion (5); the phenate ion in the hydronium-form stripper was converted to phenol, a very weak acid, which contributed little to the conductivity of the background. The most significant development in anion eluents, however, was the discovery of carbonate eluents that converted to the weak and therefore feebly conducting carbonic acid in the second column (9). Thus, carbonate eluents became widely used for anion IC for many years to come (Figure 4).

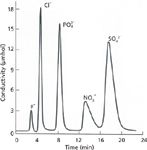
Figure 4: Status of anion IC using carbonate eluent, circa 1974.
With these developments we introduced a new vocabulary to IC; we realized that the second bed was really converting the eluent to a less conducting form rather than removing it entirely ("stripping") so suppressor and eluent suppression seemed more appropriate terms and we adopted them from then onwards.
By 1975, we had established the foundations of IC. Particularly, we had developed the procedures for producing anion exchangers in colloidal form, a keystone of stationary-phase production.
1975: IC Goes Commercial
In 1975, the Dow Chemical Company, which by this time had applied for several patents on the new technology, established a licensing agreement with Durrum Chemical, a small company whose main product was amino-acid analyzers. A separate business unit was formed within Durrum to pursue the commercialization of IC. This unit was later spun out of Durrum to become Dionex Corp., surely a major landmark in the evolution of IC. In September of 1975, Dionex signaled IC's public availability by demonstrating the first commercial instrument at the fall meeting of the American Chemical Society. It was also at this time that the term ion chromatography was used for the first time. (In 1975, the term ion chromatography referred exclusively to the combination of ion-exchange separation, eluent suppression, and conductometric detection. In later years it came to embrace a wide variety of techniques of separation and detection.)
That same fall we published the first article on the new technique (5).
Although the center of gravity of development now moved to Dionex, our small group at Dow stayed involved and had much still to contribute.
1979: IC without Suppression
Although packed-bed suppressors had enabled sensitive conductometric detection, they had some drawbacks. Notably, there was the drifting of elution times for certain analyte peaks (5,6) and a few analytes such as nitrite were degraded by interactions with the resin in the suppressor. Also, there were the interruptions for suppressor regeneration, even though we had made the interruptions to regenerate less obtrusive by arranging for the suppressor to last about an 8-h day and regenerate overnight. The picture on suppressors changed in 1979 when it was shown that ion chromatography could be accomplished using ion exchange and conductometric detection without a suppressor (10–12). This new development turned the spotlight on the suppressor and the drawback of the interruptions for its regeneration. Additionally, this new version of IC was often promoted as avoiding the "complexity" that the suppressor added. The interruptions argument was a legitimate one; the complexity argument was much less so. It is true that for samples sufficiently burdened with analyte, suppressorless IC is an adequate performer, but it has been demonstrated in practice and in theory (13) that the so-called complexity of adding a suppressor is the price of extracting the maximum sensitivity from conductometric detection. (Please note: In the context of this article, the term sensitivity describes a technique's ability to detect low levels of analyte.) This new development did, however, ignite efforts to devise better suppressors.
Continuous Suppression
When I performed the first anion separation in 1971, I used as suppressor a coil of sulfonated polyethylene tubing immersed in a stirred suspension of a cation-exchange resin (Dowex 50) in the hydronium form. While the sodium hydroxide effluent from the separator was passed through the lumen, sodium ions diffused across the wall of the tubing and exchanged with hydronium ions from the resin as its particles made bumping contacts with the exterior wall of the tubular membrane. The hydronium ions in turn diffused in the opposite direction and united with hydroxide ions to form water. These membrane devices worked quite well as a continuous suppressor but were fragile and prone to bursting, and because the bed suppressors were quite robust and we had more pressing priorities, we shelved the continuous tubular suppressor. When suppressorless IC emerged, we revived the membrane concept and succeeded in fabricating a number of more rugged devices (14,15) and they became the first in a series of continuous, chemically regenerated eluent suppressors. At about the same time, Ban and others obtained a patent on a similar continuous suppressor (16).
Dionex's 1991 introduction of a flat membrane continuous suppressor, the MicroMembrane Suppressor (MMS), was a landmark event. In this device, using anion analysis as the example, the effluent from the separator passed through the narrow channel between closely spaced, flat cation-exchange membranes in the hydronium form. The outside of the membranes was bathed by a continuous stream of sulfuric acid that supplied hydronium ions in exchange for the sodium extracted from the inter-membrane channel. These devices were very rugged, could suppress higher concentrations of eluent than their predecessors, and, with their low-volume intermembrane channels, they did not degrade the efficiency of the chromatography to an appreciable extent.
With these developments, the suppressor had evolved from being a conspicuous part of IC, and something of a bother, to being practically invisible to the user.
But further important developments in suppressors lay ahead.
Electrochemical Regeneration of Suppressors
Although the flat membrane continuous suppressor was a major advance, it still had some limitations. In the first place, it required a continuous supply of a chemical regenerant. Secondly, although the membranes were preferentially permeable to the suppressing ion, they did allow some leakage of its co-ion; in an anion suppressor, for example, some regenerant sulfuric acid leaked into the mainstream, raising the background conductivity and compromising the measurement of analytes.
As early as 1984, Jansen and others had shown that electrochemistry could be used in membrane devices to effect eluent suppression (17). By placing electrodes in the regenerant compartments of a device of MMS-like construction, ion transport across the intermembrane space was assisted by the electric potential applied to the electrodes. However, because electrolyte was used in the electrode chambers, these devices could be expected to show undesirable electrolyte leakage into the mainstream.
Another landmark in the development of suppressors was the electrochemically regenerated suppressor or the Self-Regenerating Suppressor (SRS) of Dionex (18).
While these devices used an arrangement of membranes and electrodes similar to the Jansen device and to the MMS, the electrode compartments of the SRS were flushed with deionized water. Using anion analysis with sodium hydroxide eluent as the example, when the electrodes were DC-polarized, hydronium ions produced at the anode were driven by the applied field across the cation-exchange membranes, forcing sodium ions into the cathode compartment where they united with the cathodically generated hydroxide ions and the sodium hydroxide was flushed to waste. By eliminating the chemical regenerant, the SRS eliminated the problem of regenerant leakage. And in an improved embodiment of the SRS, the water effluent from the IC operation was directed to the electrode compartments, thus eliminating the need for an extra water pump (19).
Electrochemistry also revived the packed-bed suppressor (20,21). In the Dionex Atlas suppressor, a small packed bed of ion-exchange resin, embraced by ion-exchange membranes, is continuously regenerated by polarizing the bed.
Revival of the Packed-Bed Suppressor with Chemical Regeneration
By the 1990s, analytical chemistry was augmented by a powerful ally, the computer. With the computer came the ability to automate many operations. Initially, we had introduced the "dogma" that a packed bed needed to suppress many samples before regeneration, but we now realized that with automated valve switching, a small suppressor with just single-sample capacity was viable and would require little intervention from the user (22). This basic idea was later implemented by manipulating three small suppressor beds in a clever three-compartment-revolver device (23) and marketed by Metrohm. Two major advantages of this small-bed approach over the earlier large suppressor beds are the virtual elimination of peak drifting and minimal degradation of chromatographic efficiency by peak spreading in the void space of the small suppressor bed.
Electrochemical Generation of Eluents for IC
While electrochemistry was a boon to suppression, it had another important role to play in IC.
Pioneering work by Dasgupta and others (24,25) had demonstrated that while membrane systems could remove eluent in IC they could also be used to introduce eluent in a controlled way, simply by pumping water to a suitable electrically polarized membrane device. They also recognized that the production of "pure" sodium hydroxide by such a system could provide major advantages for anion analysis by IC. In the early years of IC, carbonate eluents were successful and widely used but they had a few significant shortcomings. One was that carbonate suppressed to carbonic acid, which has low conductivity but orders of magnitude higher conductivity than pure water. Another was that the carbonate background conductivity was lowered by the presence of analyte; this was not a big issue when the analyte was abundant, but caused nonlinear responses at lower analyte levels. A third drawback was that gradient elution, as it became more of a requirement, was complicated by the ramping baseline conductivity of the carbonic acid background. Sodium hydroxide (or potassium hydroxide) as eluent had always been a sort of holy grail for IC because it could be suppressed to the ideal background, water, but two issues delayed its adoption: Hydroxide ion was a relatively ineffective displacing ion, thus demanding high suppression capacities, and it was notoriously difficult to prevent its contamination by omnipresent carbon dioxide that altered its eluting power in unpredictable ways and led to unstable backgrounds. Although the new MMS suppressors had much greater suppression capacity, thus diminishing the first issue, the carbonate-in-the-eluent problem remained. The membrane-based electrochemical generator alleviated the contamination problem to a great extent because the generator could be provided with ultrapure water and the generator was intrinsically a generator of pure carbonate-free base (26).
The new eluent generators had these positive features:
- The operator was freed from the task of frequently preparing eluents with its attendant problems of contamination and occasional operator error.
- Eluent concentration was controllable simply by controlling the current in the generator or the flow rate of the water stream, or both.
- They greatly simplified the creation of gradients.
Concurrent with these developments in hydroxide generators, new separation media with much greater affinity for hydroxide (see a later article in this issue) enhanced the impact of the electrochemical generators in anion analysis.
Ion Reflux and Eluent Recycling
While the new electrochemically based eluent generators have made a major change to the trajectory of IC, they do exhaust and have to be replaced at a cost to the user. In the late 1990s, we invented a hybrid of suppression and regeneration where the effluent from the suppressor is not discarded to waste but instead is recaptured and used again as eluent. In principle, these systems could work perpetually simply by pumping water to the IC system. In one embodiment, called ion reflux, the three components of an IC operation — eluent generation, separation, and suppression — were performed continuously within a single column, with just water as the pumped phase. In another embodiment of ion reflux the separator phase was uncoupled from the other two functions, allowing any stationary phase to be used (27,28).
Another invention, called eluent recycling (29), also enabled reuse of eluent.
Reference 30 provides a comprehensive and detailed review of these developments in electrochemistry as applied to suppression and eluent generation in IC.
Other Detection Methods in IC
Although conductometric detection was the method that launched IC, it became obvious that other detectors could be used with the new separation media. UV detectors were effective when the analytes were UV-absorbing. And of course, UV detectors did not require a suppressor, although it should be added that conductometric detection was often the preferred method when the analyte mixture contained target ions, some of which were UV-absorbing and others not.
It had been a sort of dogma in detection that UV detectors were usable only if the analytes were UV-absorbing. We showed, however, that using ion-exchange separation coupled to UV detectors could indeed be used to detect and sensitively measure UV-transparent ions (31). We called this combination indirect photometric chromatography (IPC) and the detection principle indirect photometric detection (IPD). Although IPD had eliminated the need for a suppressor and performed well in many applications (32), it lacked the sensitivity reach of conductometric detection and received little promotion in IC. However, the principle became widely used in capillary electrophoresis and somewhat in HPLC. Our work was rewarded by recognition as a milestone in analytical chemistry (33).
Carbohydrates and alcohols are not usually thought of as ionics, but Rocklin and Pohl discovered how their intrinsic ionicity could be expressed and used to chromatographically separate them (34). Because carbohydrates are extremely weak acids they express their ionicity only at very high pH, but Pohl and coworkers saw the opportunity of exploiting this and separated carbohydrates by ion exchange using strongly basic eluents (Figure 5). The anion-exchange resins of IC, with their great stability in high-pH environments, were important facilitators of this new technique. Of course, suppressed conductometric detection was a nonstarter because the carbohydrate anions would revert to their non-conducting form in the typical suppressor, so amperometric detection methods, particularly pulsed amperometric detection (PAD) (35,36), became important adjuncts to IC and enabled IC separation of a wide variety of analytes (37).

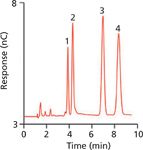
Figure 5: Determination of carbohydrates by IC: 1 = glucose, 2 = fructose, 3 = lactose (internal standard), and 4 = sucrose. Adapted from reference 43 with permission.
This application of IC to nonionics was a distinctly new and different trajectory for the method and certainly a landmark event.
IC with Water as Eluent
Although in the early years we made little progress using simply water as the eluent, others have been notably more successful. A significant landmark in the evolution of IC is the work of Lamb and others using macrocylic species that form selective and reversible complexes with electrolytes in pure water (38).
Applications of IC
It is impossible, in an article of this length, to do justice to the pioneer users of IC and the many landmark applications that expanded the method's usefulness. Therefore, I will restrict my choice to two: the first symposium on IC and the series of developments that have produced astounding advances in the detectability limits of IC. (References 32 and 37 are recommended for accounts of the myriad applications of IC.)
After the introduction of IC in 1975, it took a certain boldness to embrace this brand-new technology; there were many who said it would not prosper, or as one guru expressed it — so I've been told — "IC was fatally flawed" by the suppressor. But many did embrace it, and some of their early creative applications are recorded in the proceedings of that first symposium at Gatlinburg organized by the U.S. Environmental Protection Agency (39).
There are at least two industries that depend on water of the highest available purity: the power-generating industry and the electronics industry. The first must protect its critically important boilers from the corrosive effects of ions, notably chloride, while in the second, sensitive electronic components can be irreparably damaged by traces of electrolytes in processing water. Careful monitoring of water streams is vital to both these industries so there is great demand to extend the sensitivity of IC as much as possible. By adhering to scrupulously clean procedures, such as the in situ production of ultrapure water by electrochemical deionizers, adventitious contamination of samples and eluents can be avoided and techniques have been developed that enable the detection and measurement of ions at the parts-per-trillion level (Figure 6).

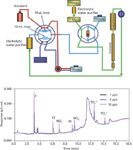
Figure 6: A dual-loop injection valve and concentrator column are used to automate calibration and sample loading. Electrolytic deionization of the conductivity cell waste produces ionically pure water for in-line standard dilution, sample loading, and loop rinsing. This system results in extremely low detection limits (parts per trillion), low blanks, and high precision. The chromatogram was generated using a Dionex ICS3000 system with a KOH generator and ASRS300 suppressor (2 mm). Columns: IonPac AG17 and AS17 (2 mm); flow rate: 0.5 mL/min; loop volumes: 10 µL and 10 mL. Concentrator column: UTAC-ULP2 low pressure anion concentrator. (Adapted with permission from Trovion Singapore Pte Ltd.)
In our earliest embodiments of IC we were proud of our ability to measure at the parts-per-million level; all the efforts that have extended detectability of ions by a million-fold are truly landmarks in the evolution of IC.
Speculations on the Future of IC
It is to be expected that IC will continue its penetration into ion analysis and it is likely that special cases will emerge, like sulfate in the early days or perchlorate more recently, where IC will display its unique ability to solve urgent analytical problems.
As to the future of IC technology, the indicators already point in the direction of smaller-scale instruments, with their many benefits (40). From our earliest days in IC we were aware that the conductivity cell was unique among chromatographic detectors in its amenability to miniaturization and recent developments in detectors, where electrodes make capacitive contact with the contents of capillaries (41), are a significant step in this direction.
IC and other forms of liquid chromatography use elution times of analytes as the defining measure of their chromatographic behavior. The elution time of an analyte is not a fundamental property in chromatography, but time has the great advantages of ease of measurement and of almost indefinite subdivision. However, for elution time to be a reliable definer of the chromatogram, the chromatographic pump must deliver very stable and precisely controlled flow. The pump is thus a flowmeter as well as a mover of eluent. As a result, the pump is often the most highly engineered and costly component in the system. Further, reductions in pump size do not seem to be keeping pace with the miniaturization of stationary phases and detectors.
A more fundamental property than an analyte's elution time is its elution volume, and development of volume flow meters as a separate chromatographic device would relieve the pump of this task. This should, in turn, lead to a significant reduction in pump size and complexity. We have recently taken a step toward reducing pump size by inventing an electrochemically driven pump (42).
In IC there is a property of analyte elution that is even more fundamental than elution volume. Assuming that the eluent and the stationary phase have been established, then, with one important caveat, the number of equivalents of displacing ion required to elute an analyte is fixed; or the number of coulombs to elute is fixed if an eluent generator is being used. (This is true only if the analyte and the displacing ion are of the same valence; it is more complicated if they are not, but there are solutions for this complication.) So IC chromatograms might be defined not by time or volume but by the number of coulombs applied. Does this mean not only that the pump need not be supremely stable but that we might also in some cases even dispense with a flow meter? In IC, the field is open for innovation in how eluent is delivered and its flow measured.
IC eluent generators include a reservoir of concentrated acid or base as the source of the eluent and the membrane or membranes that separate this reservoir from the mainstream must be of substantial thickness to prevent leakage of the base (or acid) into the mainstream. A reservoir of ion-exchange resin, the perfectly nondiffusible electrolyte, would offer a remedy for this problem and is worth examining. And the neglected area of eluent recycling (27–29) is likely to be re-examined in the years ahead.
In the next several years, the landscape of IC will change as dominant patents expire and other players enter the field. Where this might lead is beyond speculation, but we can be certain that new inventions and innovations will emerge to challenge the status quo, just as they have since the beginning of IC.
Hamish Small was born and received his early education in Northern Ireland where he received B.Sc. and M.Sc. degrees from the Queen's University of Belfast. He worked from 1949 to 1955 for the United Kingdom Atomic Energy Authority at Harwell in England before immigrating to the United States and joining the Dow Chemical Company in Midland, Michigan. He remained at Dow until 1983, before retiring to pursue his career in independent research and invention. He has maintained a close association with Dionex since its inception. Small is credited with 49 U.S. patents, several of which cover key inventions in ion chromatography. Direct correspondence to: hamishsmall29@gmail.com


Hamish Small
References
(1) H. Small, Chem. Heritage 21(3), 10–11 and 38–41 (2003).
(2) H. Small, J. Chem. Educ. 81(9), 1277 (2004).
(3) J. Inczedy, Analytical Applications of Ion Exchangers (Pergamon Press, Oxford, 1966).
(4) S. Moore and W.H. Stein, J. Biol. Chem. 192, 663–681 (1951).
(5) H. Small, T.S. Stevens, and W.C. Bauman Anal. Chem. 75, 1801–1809 (1975).
(6) H. Small, Ion Chromatography (Plenum Publishing, New York, 1989).
(7) H. Small and W.C. Bauman, US Patent 3,920,397 (1975).
(8) H. Small and T.S. Stevens, U.S. Patent 4,101,460 (1978).
(9) H. Small and J. Solc, "Ion Chromatography – Principles and Applications" Proceedings of An International Conference on "The Theory and Practice of Ion Exchange", Society of Chemical Industry, London (1976).
(10) K. Harrison and D. Burge, Pittsburgh Conference on Analytical Chemistry, Abstract #301 (1979).
(11) D.T Gjerde, J. Fritz, and G. Schmuckler, J. Chrom. 186, 509–519 (1979).
(12) J.S. Fritz, D.T. Gjerde, and C. Pohlandt, Ion Chromatography (Dr. Alfred Huthig, Heidelberg, 1982).
(13) H. Small, Ion Chromatography (Plenum Publishing, New York, 1989), pp. 180–186.
(14) T.S. Stevens, J.C. Davis, and H. Small, Anal. Chem. 53, 1488–1492 (1981).
(15) T.S. Stevens, J.C. Davis, and H. Small, U.S. Patent 4,474,664 (1984).
(16) T. Ban, T. Muryama, S. Muramota, and Y. Hanaoka, U.S. Patent 4,403,039 (1983).
(17) K.H. Jansen, K.H. Fisher, and B. Wolf, U.S. Patent 4,459,357 (1984).
(18) C. Pohl, R. Slingsby, J. Stillian, and R. Gajek, U.S. Patent 4,999,098 (1991).
(19) J.R. Stillian, V.B. Barreto, K.A. Friedman, S.B. Rabin, and M. Toofan, U.S. Patent 5,248,426 (1993).
(20) H. Small, Y. Liu, J.M. Riviello, N. Avdalovic, and K. Srinivasan, U.S. Patent 6,325,976 (2001).
(21) J.M. Anderson, R. Saari-Nordhaus, B.C. Benedict, C. Sims, Y. Gurner, and H.A. Pham, U.S. Patent 6,468,804 (2002)
(22) H. Small, J. Riviello, and C. Pohl, U.S. Patent 5,597,734 (1997)
(23) H. Schafer, M. Laubli, and P. Zahner, U.S. Patent 6,153,101 (2000).
(24) D.L. Strong, P.K. Dasgupta, K. Friedman, and J.R. Stillian, Anal. Chem. 63, 480–486 (1991).
(25) P.K. Dasgupta, D.L. Strong, J.R. Stillian, and K.A. Friedman, U.S. Patent 5,045,204 (1991).
(26) Y. Liu, H. Small, and N. Avdalovic, U.S. Patent 6,225,129 (2001).
(27) H. Small and J. Riviello, Anal. Chem. 70(11), 2205–2212 (1998).
(28) H. Small, U.S. Patent 5,914,025 (1999)
(29) H. Small, Y. Liu, and N. Avdalovic, Anal. Chem. 70(17), 3629–3635 (1998).
(30) Y. Liu, K. Srinivasan, C. Pohl, and N. Avdalovic, J. Biochem. Biophys. Methods 60, 205–232 (2004).
(31) H. Small and T.E. Miller, Anal. Chem. 54, 462–469 (1982); U.S. Patent 4,414,842 (1983).
(32) P.R. Haddad and P.E. Jackson, Ion Chromatography: Principles and Applications (Elsevier, Amsterdam, 1990).
(33) Milestones in Analytical Chemistry (American Chemical Society, Washington DC, 1994).
(34) R.D. Rocklin and C.A. Pohl, J. Liq. Chrom. 6, 1577 (1983).
(35) S. Hughes and D.C. Johnson, Anal. Chim. Acta 149, 1–10 (1983)
(36) T.R. Cataldi, C. Campa, and G.E. Benedetto Fresenius, J. Anal. Chem. 368, 739–758 (2000)
(37) J. Weiss, Ion Chromatography, second edition (VCH, Weinheim, 1995).
(38) J.D. Lamb and J.S. Gardner, Macrocyclic Chemistry: Current Trends and Future Perspectives, Karsten Gloe, Ed. (Kluwer Academic Publishers, Dordrecht, the Netherlands, 2005), pp. 349–363.
(39) E. Sawicki, J.D. Mulik, and E. Wittgenstein, Eds., Chromatographic Analysis of Environmental Pollutants (Ann Arbor Science, Ann Arbor, Michigan, 1978).
(40) Y. Liu, V.M.B. Barreto, C.A. Pohl, and N. Avdalovic, Patent Application # 20120228227 (2012).
(41) J.A.F. da Silva and C.L. do Lago. Anal. Chem. 70, 4339–4343 (1998).
(42) H. Small, Y. Liu, and C.A. Pohl, U.S. Patent 8,133,373 (2012).
(43) Dionex Application note #92 (ThermoFisher Scientific, Sunnyvale, California).

















. | Image Credit: © Joan Vadell - stock.adobe.com")
Detecting Hyper-Fast Chromatographic Peaks Using Ion Mobility Spectrometry
May 6th 2025Ion mobility spectrometers can detect trace compounds quickly, though they can face various issues with detecting certain peaks. University of Hannover scientists created a new system for resolving hyper-fast gas chromatography (GC) peaks.


. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

Sorbonne Researchers Develop Miniaturized GC Detector for VOC Analysis
April 16th 2025A team of scientists from the Paris university developed and optimized MAVERIC, a miniaturized and autonomous gas chromatography (GC) system coupled to a nano-gravimetric detector (NGD) based on a NEMS (nano-electromechanical-system) resonator.


