Translations Between Differing Liquid Chromatography Formats: Advantages, Principles, and Possible Pitfalls
The numerous advantages of translating gradient chromatographic methods between the differing formats of liquid chromatography (LC) have been explored and discussed.
The numerous advantages of translating gradient chromatographic methods between the differing formats of liquid chromatography (LC) have been explored and discussed. Although translations in principle obey well-defined chromatographic theories, the authors investigate a number of potential pitfalls that may result in poor translations as exhibited by selectivity differences, changes in efficiency, and hence failure to meet resolution system suitability criteria. The consequences of these pitfalls are examined and the regulatory implications of method translation are explored.
As a result of the introduction of commercially available sub-2-µm porous particles (1), sub-2-µm, 3-µm, and 5-µm superficially porous (2,3) particles, and ultrahigh-pressure liquid chromatography (UHPLC) instrumentation (4,5) from 2004 onward, there has been an increasing interest in the ability to perform accurate translations between different liquid chromatography (LC) formats. An example would be translating between 150 mm × 4.6 mm, 5-µm dp formats on standard high performance liquid chromatography (HPLC) systems and 50 mm × 2.1 mm, sub-2-µm formats on UHPLC systems while maintaining the same resolution. The findings of a recent survey of major chromatographic users predicted that the use of standard HPLC systems is expected to steadily decline from 2011 to 2015 with a concomitantly higher usage and purchase of UHPLC systems predicted over the same time frame (6).
There are a plethora of reasons for this shift in LC format usage and purchase, all of which are based on sound chromatographic theory (5,7,8). From the extensive experience of the authors within the pharmaceutical industry, the major drivers for this shift appear to be increased productivity (that is, reduced analysis time) coupled with minimal loss of information quality or an increased quality of data with no loss of productivity.
Advantages and Drivers
Increased Resolution
Reduction of the packing material particle size by a factor of two (that is, substitution of 3–3.5 µm particles by 1.7-µm particles), while keeping other operation factors constant, should result in an increase of resolution of approximately 30–40%.
Speed of Analysis
A reduction in column length (L) and particle size (dp) while keeping the L/dp ratio constant (for example, substitution of a 150-mm column with 3–3.5 µm particles for a 75-mm column with 1.7-µm particles) should maintain the same chromatographic efficiency and hence resolution, while reducing the gradient analysis time by 50% (same velocity typically used for large molecules) to 70% (higher velocity typically used for small molecules) and substantially increasing productivity. This approach is vitally important for the analysis of increasingly larger numbers of samples (that is, to better describe a process or formulation performance), increased utilization of instruments, the analysis of labile samples, and rapid at-line analysis (that is, process analytical technology).
A compromise between the approaches of increased resolution and speed of analysis has been the use of 100-mm columns with sub-2-µm particles, which results in a 60% reduction in gradient time and an approximate 10% increase in resolution for small molecules.
Reduced Solvent Consumption
Converting a standard HPLC method that uses a 150 mm × 4.6 mm, 3–3.5 µm column to a 100 mm × 2.1 mm, 1.7-µm or 100 mm × 1.0 mm, 1.7-µm column in theory offers a possible reduction in solvent consumption of approximately 86% to 97%. In practice, it is less often because of the necessity to prime the LC lines. During a global implementation of UHPLC within AstraZeneca (during the period of 2007–2010) involving 41 UHPLC systems, a reduction in solvent consumption of 63% was realized compared to the theoretical reduction of 77% (9).
Ease of Method Transfer
Within many industries it is often standard practice to transfer the chromatographic testing from the research and development (R&D) laboratory to a contract research organization (CRO), operation, or quality control (QC) sites. This method transfer exercise can be made even more problematic because it is now quite common for many R&D departments to develop only UHPLC methods. However, not all receiving laboratories have sufficient UHPLC capacity or experience and, thus, method translations become necessary. The reverse of this is becoming true in that QC laboratories, which have moved predominantly to UHPLC, may have to use UHPLC methods for the analysis of legacy products or methods that use HPLC columns.
Increased Instrument Utilization
The drive for increased productivity and efficiency has necessitated an increased flexibility and utilization of available instrumentation. Many companies have a rolling program to replace their worn out HPLC systems with a reduced number of UHPLC systems capable of running LC methods based on both 5-µm and sub-2-µm particles. In addition, valve arrangements are used that allow queuing of both HPLC and UHPLC methods on the same LC system, hence a reduced number of LC systems allow continuous operation.
Principles of Method Translation
Because many translation guides successfully describe translations of isocratic LC methodologies (10), this article will focus entirely on the translation of gradient LC methodologies, which can be more difficult to perform. In theory, the translation required to maintain the chromatographic selectivity and performance of either an isocratic or gradient separation is very simple (7,11). The additional consideration that has to be accounted for in gradient separations is that a constant ratio must be maintained between the volume of each segment in the gradient over the column dead volume (8,12–15). After the introduction of sub-2-µm particles and UHPLC in the mid-2000s, several articles were published that focused exclusively on translations between 4.6- and 2.1-mm i.d. columns (7,8,10,16–18). To assist the practicing chromatographer in all types of translations, several commercial and academic computer applications have been developed (19); however, although these computer applications undoubtedly aid chromatographers, they all possess certain drawbacks to successful method translation.
In contrast to previous publications, this article only briefly describes the underlying theory and equations relating to chromatographic method translation principles (readers are encouraged to see the sidebar "Theory for Translations" for more information). Instead, this article will focus on how to perform successful translations of gradient chromatographic methods between HPLC and UHPLC systems, the accuracy that can be expected, and the potential pitfalls that chromatographers must be aware of and how to successfully avoid them. If the necessary precautions are taken, it is the experience of the authors that gradient translations work extremely well. For example, 11 LC methods were translated to or from UHPLC within AstraZeneca; the maximum deviations in relative retention times were only between 0.02 and 0.05, which was deemed acceptable (9,18).



Theory for Translations
Experimental
Experimental work was performed on Agilent 1100, 1260, and 1290 LC systems of which the dwell volume and system volumes had been previously well characterized. Any experimental data including the injector programs used can be supplied by the authors on request. For the delayed injection work on the Agilent LC systems, the injector must be in the bypass mode (that is, no flow through the injector). After the calculated delay time, the flow was returned to the mainpass position (that is, flow through the injector), then after a predefined time (time = [5 × [injection volume + 5]]/flow rate) to flush the sample onto the column, the flow was returned to the bypass position. At the top of the gradient the flow was returned to the mainpass position to wash the injector with the mobile phase to avoid carryover and subsequently flush the injector with the starting mobile phase composition prior to the next injection. Zorbax Eclipse XDB C18 columns (Agilent Technologies) of 1.8-, 3.5-, and 5-µm particle sizes were selected for the small molecule work because they exhibit a good scalability with respect to efficiency and particle size (R2 = 0.9 for N versus 1/dp) (20).
Translations were made using a Microsoft Excel spreadsheet and the equations described in the sidebar "Theory for Translations." A corresponding software application for these types of translations is now freely accessible at the ACD/Labs website (21). In addition, a more complete set of translation tools will be included in version 2014 of the ACD/Chrom Workbook software (22).
Potential Pitfalls in Method Translations
Observed Selectivity Anomalies as a Result of Differences in Dwell Volume
The system dwell volume (VD) is defined as the volume from the point where the two solvents A and B first meet to the inlet of the column. For accurate translation of gradient methods it is of critical importance that the ratio between the dwell and dead volume (VM) is kept constant; that is, the difference in ratio between the system dwell volume and the column dead volume needs to be negligible (Δ = [VD/VM]original – [VD/VM]new must approach 0). If this is not the case, then differences in chromatographic selectivity and relative retention time shifts may be observed. One such example is shown in Figure 1, where a series of analgesic related drugs have been chromatographed on the same 50 mm × 2.1 mm, 1.8-µm dp column using a binary high-pressure mixing system with low dwell volume (that is, 202 µL) and a quaternary low-pressure mixing system with a higher dwell volume (that is, 990 µL). Note that in this example a translation from a system with VD/VM = 1.8 (Figure 1a) to a system with VD/VM = 9 (Figure 1b) resulted in coelution (peaks 7 and 8) as well as a reversal of elution order (peaks 3 and 4).

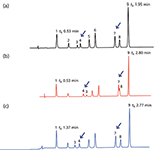
Figure 1: An illustration of how a larger VD/VM for the translated method can be compensated using a delayed sample injection (Î negative). The data also illustrates how VD/VM differences can affect the selectivity. The chromatograms have been scaled by alignment of the first and last eluted peaks. (a) binary model 1290 Agilent system, VD/VM = 1.8, injection in mainpass; (b) quaternary model 1290 Agilent system, no correction, VD/VM = 9.0, injection in mainpass; (c) quaternary model 1290 Agilent system, 0.93-min injection delay, VD/VM = 8.2, injection in bypass. Other conditions for (a)â(c): column: 50 mm à 2.1 mm, 1.8-µm dp; flow rate: 0.75 mL/min; mobile-phase A = 0.1% formic acid in water; mobile-phase B = 0.1% formic acid in acetonitrile; gradient: 5â27% B in 2 min; temperature: 40 °C. Peaks: 1 = paracetamol, 2 = 4-hydroxybenzoic acid, 3 = caffeine, 4 = 3-hydroxybenzoic acid, 5 = salicylamide, 6 = acetanilide, 7 = aspirin, 8 = salicylic acid, 9 = phenacetin.
To compensate for a larger VD/VM on the translated method (that is, Δ = [VD/VM]original – [VD/VM]new = negative), the gradient must be started before injecting the sample — that is, so-called injection delay or pre-injection volume — which can be easily implemented on certain LC systems (such as the Agilent 1290 or the Waters Acquity H-class systems). Equations 9 and 10 in the sidebar describe this process in detail. It should be noted that it is only necessary to determine the VD (23) once for a certain LC configuration. Figure 1 illustrates how the injection delay principle can be used to compensate for VD differences and thereby maintain the original selectivity and relative retention (Figure 1a versus 1c).
To compensate for a smaller VD/VM on the translated method (that is, Δ = [VD/VM]original – [VD/VM]new = positive) the translated gradient must possess an isocratic hold before commencing the gradient.
A small VD/VM results in a gradient with sharp changes (a Z-shaped gradient), whereas larger VD/VM will result in smoother, more S-shaped changes. This difference in gradient shape may also result in selectivity changes, mainly for fast separations on short columns (for example, 2-min gradients on 50 mm × 2.1 mm columns). To address this problem with possible selectivity changes, Agilent Technologies developed an approach (24) in which the company's model 1290 UHPLC systems can mimic the gradient shape of systems with a larger system dwell volume.
Observed Selectivity Anomalies as a Result of Incorrect Dead Volume Estimation
The column dead volume is a fundamental parameter that has a significant impact on translations and their subsequent accuracy (equations 1, 9–13 in the sidebar). To the best of our knowledge, all available translation applications erroneously assume an equal porosity for all stationary phases irrespective of their nature (equation 2 in the sidebar). In cases where this assumption is not valid, significant errors in translated gradient times are possible. For example, large differences in porosity and hence dead volume can be expected and observed between porous and superficially porous particles (that is, 10–29% difference was observed for seven different types of superficially porous columns [25] and 13–37% predicted difference according to equations 2–5 in the sidebar). Consequently, translated gradient times may have an error of the same magnitude; for nonporous particles the error will be even greater (that is, 52%).
Equations 2–5 in the sidebar describe how VM can be estimated for columns based on superficially porous particles using their reported particle and core diameters. However, these and other VM estimates are all associated with an error that can be quite substantial. The use of quoted particle sizes may be misleading as there are many differing ways that particle size can be determined and reported (20). The pore size of the particles also affects the porosity (100-Å columns have a porosity of ~0.60, whereas 300-Å columns have a porosity of ~0.75). VM estimations may also be inaccurate because of the fact that columns packed with different particle sizes are often packed with different pressures, which can result in lower porosities for columns designed for UHPLC. Consequently, for columns packed with porous particles, the difference between theoretical and measured dead volumes can be quite substantial. A comparison of 14 brands of modern columns from various vendors packed with porous particles exhibited a dead volume prediction error of ±28% (25). This value corresponds to an error in gradient time of up to 28%. Figure 2 exemplifies how a 29% error in VM can affect retention time as well as chromatographic selectivity.

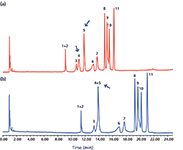
Figure 2: An illustration of how a translation from one column to another may affect both retention and selectivity if the translation is incorrectly performed (that is, assuming equal porosity between materials). Conditions: (a) Column: 150 mm à 2.1 mm,3.6-µm dp, 3.2 µm dcore, VM = 0.27 mL, 200-à superficially porous particle Aeris widepore XB-C18; flow rate: 0.3 mL/min; gradient: 27â65% B in 30 min; mobile-phase A: 0.1% trifluoroacetic acid in water; mobile-phase B: 0.08% trifluoroacetic acid in acetonitrile; temperature: 40 °C. Conditions (b): Gradient scaled assuming the same porosity as for 300 à porous particles, that is, VM = 0.38 mL and a gradient time of 41.8 min. Other conditions same as (a). Sample: proteins with pI ranging from 3.6 to 9.3 (Sigma-Aldrich I3018 IEF mix 3.6â9.3).
Figure 2a demonstrates the experimental chromatogram derived by using a 200-Å superficially porous material with a dp and dcore of 3.6 µm and 3.2 µm, respectively, with a measured VM for the column of 0.27 mL. In comparison, when the methodology is translated using the same column (to eliminate any stationary-phase selectivity differences), but assuming that it is 300 Å and fully porous in nature, the VM is calculated as 0.38 mL and the new gradient yields a significant change in selectivity (see Figure 2b).
Given the above discussion, the authors strongly recommend that VM values are experimentally determined to obtain accurate translations. VM can be easily determined by injecting a reversed-phase-LC dead time marker such as uracil or thiourea.
Observed Selectivity and Peak Width Anomalies as a Result of Differences in Column Thermostat Design Between LC Instrumentation
Differences in column thermostat design used by differing LC instrument vendors will result in different temperatures being achieved within the column despite identical "set-point and feedback" temperatures being recorded in the column compartment. The extent of this temperature deviation is dependent on instrument design, flow rate, and temperature set point. According to our experience, deviations of 5 °C or more are commonplace (25). This degree of temperature difference is sufficient to cause significant selectivity differences when transferring a method from one type of LC instrument to another. Consequently, when translating from HPLC to UHPLC or transferring a method from one type of HPLC system to another, it is very likely that observed selectivity differences are related to differences in column temperature.
Column thermostat design affects not only selectivity, but also efficiency. It has been found that a precolumn heat exchanger in combination with a still air column compartment can reduce radial temperature gradients in the column and thereby results in narrower peaks compared to a water bath or a thermostat based on a forced air flow principle (26,27).
To address this problem, we recommend that the system suitability test (SST) section of the method describes how the resolution is affected by temperature and how corrections can be employed; this description can be achieved, for example, by illustrating chromatograms corresponding to 30 °C, 35 °C, and 40 °C for a method developed on an instrument at 35 °C or by the construction of van't Hoff plots (that is, log k versus 1/temperature). By following this procedure, users can see in what direction and approximately how much the temperature needs to be changed to meet the SST acceptance criteria. This approach is supported by the United States, European, and Japanese pharmacopeias in that they allow a ±5 °C adjustment of temperature to obtain the right selectivity (23,40,44).
Observed Selectivity Anomalies as a Result of Differences in Heat of Friction
When the mobile phase is depressurized in the column, frictional heat is generated, resulting in an axial temperature gradient. The heat generated is proportional to both the pressure drop over the column, but also the flow has an impact on heat generation and dissipation (28,29). On a 2.1-mm i.d. column operated at a pressure close to 1000 bar, the difference between the column outlet and inlet temperature can be on the order of 5–10 °C for a 150-mm column and 10–18 °C for a 50-mm column (28,29).
Selectivity differences caused by heat of friction can be compensated for by increasing the temperature when translating from UHPLC to HPLC and thereby compensate for the reduced heat of friction. Alternatively, the temperature is decreased to compensate for an increased heat of friction when translating from HPLC to UHPLC. One approach that is recommended is to change the temperature ±2 °C, 4 °C, 6 °C, and 8 °C and thereby investigate if the observed change in selectivity is related to heat of friction and can be compensated for (18).
Observed Selectivity Anomalies as a Result of Differences in Pressure
A change in pressure affects the molar volume of solvated analytes as well as their degree of ionization (30–34). This effect typically results in an increased retention as pressure increases; however, there are exceptions to this rule (34). Differing degrees of pressure-induced retention changes can result in either enhanced or reduced chromatographic resolution. It has been reported that proteins are more sensitive to pressure-induced retention effects than lower-molecular-weight analytes because their secondary and tertiary structures are additionally affected by pressure and flow (35).
The majority of previous studies on pressure-induced retention effects have been conducted with a postcolumn restrictor to increase the pressure while maintaining the same flow rate and do not reflect typical chromatographic conditions. Furthermore, it has been suggested that heat of friction should counteract pressure-related retention and selectivity changes to some extent (28,35). Nevertheless, pressure-related selectivity differences are also seen during typical chromatographic conditions and for quite modest differences in pressure (for example, 142 bar) (36). Figure 3 illustrates a typical example of a translation from an HPLC method based on a 150 mm × 4.6 mm, 5-µm column (Figure 3a) to a UHPLC method based on a 50 mm × 2.1 mm, 1.8-µm column (Figure 3c) where a pressure increase of only 125 bar resulted in a significant change in selectivity (peaks 3 and 4). Unfortunately, it is difficult to compensate for these types of pressure effects. While it does not provide a solution, adding a postcolumn restrictor can enable confirmation that pressure is the cause of the problem (Figure 3d). To compensate for pressure-related selectivity changes, it is probably necessary to reoptimize the method by adjustments of parameters such as gradient shape and temperature.

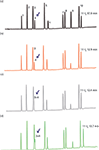
Figure 3: A translation while maintaining constant linear velocity from an HPLC method based on (a) a 150 mm à 4.6 mm, 5-µm column to the corresponding UHPLC method based on a 50 mm à 2.1 mm à 1.8 µm column (b) without and (c) with dwell volume compensation. The chromatograms have been scaled by alignment of the first and last eluted peaks. (a) binary Agilent 1100 system, VD/VM = 0.8 (injection in mainpass), 103 bar; (b) binary Agilent 1290 system, no correction, VD/VM = 1.9 (injection in mainpass), 231 bar; (c) binary Agilent 1290 system, 0.12 min injection delay, VD/VM = 1.0 (injection in bypass), 228 bar; (d) as in (b) but with a postcolumn restrictor, 714 bar. Conditions (a): Flow rate: 1 mL/min; gradient: 5â100% B in 40 min; mobile-phase A: 20 mM monobasic potassium phosphate (pH 2.7) in water; mobile-phase B: 20 mM monobasic potassium phosphate (pH 2.7) in 65:35 (v/v) acetonitrileâwater; temperature: 40 °C. Conditions for (b), (c), and (d) are the same as (a) except for a gradient time of 13.3 min, a flow of 0.208 mL/min. Peaks: 1 = terbutaline, 2 = N-cetylprocainamide, 3 = phenol, 4 = eserine, 5 = quinoxaline, 6 = quinine, 7 = ARD12495, 8 = diphenhydramine, 9 = carvediol, 10 = amitriptyline, 11 = reserpine.
Poor Efficiency Because of Differences in Extracolumn Band Broadening Between LC Instrumentation
Efficiency is dependent on retention as well as the ratio between column dead volume and the volume of the peak (equation 14 in the sidebar). Since the dead volume of an HPLC column is typically much larger than that of an UHPLC column, it is necessary to reduce the UHPLC instrument's contribution to peak volume significantly (7). This extracolumn band broadening (ECBB) — usually measured as 4σ — is typically in the order of 30–50 µL and 10 µL for HPLC and UHPLC, respectively. Even the low volume associated with modern UHPLC instrumentation has been found to be insufficiently low to compensate completely for the extracolumn band broadening contribution associated with peaks of low retention (7,37). This effect is illustrated in Figure 4, where an isocratic translation from HPLC to UHPLC exhibited the expected efficiency for late eluted peaks; however, the efficiency observed for early eluted peaks was significantly lower. Figure 5 illustrates the efficiency versus the retention factor for the same separation for other combinations of column and LC system ECBB volumes. In this example as well as in reference 7, the expected plate numbers are not reached until a retention factor of approximately 4 is reached when translating to a 50 mm × 2.1 mm column operated on an UHPLC system.

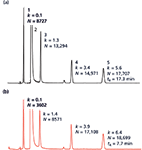
Figure 4: An isocratic translation from (a) a 250 mm à 4.6 mm, 5-µm column and a binary Agilent 1100 HPLC system to (b) a 100 mm à 2.1 mm, 1.8-µm column and a quaternary Agilent 1290 UHPLC system, showing how ECBB becomes critical for peaks with low retention when using columns with small dead volumes. The chromatograms have been scaled by alignment of the first and last eluted peaks. Flow rate (a): 1.0 mL/min; flow rate (b): 0.21 mL/min; mobile phase: 2:400:600 (v/v/v) phosphoric acid 85% w/vâacetonitrileâwater; temperature: 22 °C. Peaks: 1 = 4-hydroxybenzoic acid, 2 = acetylsalicyclic acid, 3 = salicylic acid, 4 = acetylsalicylsalicylic acid, 5 = salsalate.
Modifying the ECBB of an LC system is usually quite difficult (38). A more realistic alternative is to increase the column internal diameter. Today, 3-mm i.d. UHPLC columns are commercially available, and they can significantly reduce this problem. The drawback to this approach is that a flow rate twice as high as for a 2.1-mm i.d. column must be used, resulting in a reduced saving in solvent consumption and a potential for increased heat of friction, which could cause band broadening and selectivity differences (29).

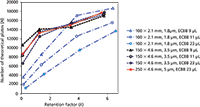
Figure 5: Number of theoretical plates versus retention factor for the separations shown in Figure 4 and for five additional combinations of columns and LC systems with different ECBB contributions.
In general, the minimized ECBB associated with UHPLC systems is advantageous; however, there is a possibility that it may cause significant band broadening when performing HPLC methods with the sample dissolved in a high proportion of organic solvent compared to the initial mobile phase. This may appear contradictory, but it can be explained by the narrower capillaries on the UHPLC system reducing the mixing between the sample and the mobile phase and thereby reducing peak focusing of the sample on top of the column. The solution to this problem is to reduce the sample volume or the amount of organic solvent in the sample solution. Alternatively, the capillary between the injection valve and the column can be replaced with a large internal diameter capillary to increase the mixing.
Differences in Linearity, Response, or Repeatability Related to Injector Design
Different LC systems possess differing injection flow principles and materials in their autosampler construction. For example, in a loop injector the sample is typically exposed to larger surface areas and other materials than in a flow through needle injector design. This larger surface area may result in a more pronounced adsorption and, consequently, also a more pronounced nonlinear response at low concentrations for loop injectors.
Other potential problems can be related to differences in the internal diameter of the injector needle and capillaries. UHPLC systems have significantly narrower injector needle and capillary diameters, which can result in poor injection repeatability because of bubble formation if the draw speed is set too high in relation to the viscosity of the sample. Another related problem is that differences in viscosity between samples and standards because of matrix differences may result in differing amounts being injected, thereby the sample concentration can be under or over-estimated.
Consequently, it is important to validate linearity, response, and repeatability of the LC method when transferring a method from one type of LC system to another.
Differences in Peak Asymmetry Related to Differences in Efficiency
The transfer of HPLC methodology to UHPLC when overloaded peaks are involved results in more-pronounced peak asymmetries being observed even if injection volumes have been correctly scaled against column volume. The explanation for this phenomenon is that the higher efficiency associated with the UHPLC method results in higher concentrations of the analyte at the apex of the peak and thus a higher degree of overloading is observed (39). Despite this overloading, the resolution of peaks adjacent to the overloaded peak is typically higher in the UHPLC method than in the HPLC method. It is possible to compensate for this overload effect by simply scaling the injection volume against column dead volumes as well as the isocratic efficiencies (equations 12 and 13 in the sidebar).
Other Issues
In the early days of UHPLC, selectivity differences were commonly observed between columns of nominally the same material but differing particle size. These differences were impossible to explain by differences in the heat of friction or pressure differences (20). It was assumed that the base silica of the smaller particle size material was subtly different compared to its larger particle size counterparts. Today this difference is less of a problem. However, if a selectivity difference is observed that cannot be compensated for by either increasing or decreasing the temperature by a few degrees to mimic heat of friction or by adding a postcolumn restrictor to mimic a pressure-induced retention change, it is more than likely that it is related to subtle differences in the heterogeneity of the base silica used.
Regulatory Aspects
The degree of revalidation that is required for a translated method depends on the intended purpose for that method and where in the R&D process it is to be used.
For pharmacopeia methods, LC translations can, in principle, be made without any formal validation exercise being performed provided that the operating ranges described in the appropriate monographs (23,40,44) are not exceeded. Unfortunately, the current operating ranges limit the ability to translate from HPLC to UHPLC. For example, the particle size can only be reduced by 50% which prevents translation from a 5-µm material to a sub-2-µm material. It is, however, expected that the pharmacopoeias will be updated in the near future to allow for such translations to take place as long as the specified selectivity and efficiency is maintained (that is, by maintaining or increasing the ratio between column length and particle diameter [41]). Hopefully, this will also result in increased regulatory flexibility in general.
For original pharmaceutical products it is necessary to validate all versions of the method that are used for analysis of samples to be used in toxicological or clinical studies. Do both the HPLC and the UHPLC methods require a full International Conference on Harmonisation (ICH) validation (42)? From a scientific point of view, it should be sufficient to submit a full ICH validation for either the HPLC or the UHPLC version of the method. For the other version, it should be sufficient to submit a minimalistic validation report claiming that the translation has been made according to first principles (that is, fundamental chromatographic theory). In principle, it should be enough to validate selectivity and linearity for the translated version of the method. It is advisable, at least during this transition period, to perform a full ICH validation and be prepared to submit the "missing" parts of the validation if requested by the authorities. It may also be prudent to include both the HPLC and UHPLC versions of the method in the experimental design used for validation of intermediate precision.
Postsubmission changes to methods are possible according to both the European Medicines Agency (EMA) and the Food and Drug Administration (FDA) regulations. However, it is uncertain what is required in the rest of the world. In addition, the cost associated with a postapproval change is often considered prohibitive.
Conclusions
This article demonstrates how the practical advantages of translating existing HPLC methodologies to those using newer column formats and UHPLC instrumentation can be realized. Advantages include increased resolution, enhanced speed of analysis, reduced solvent consumption, more efficient utilization of LC equipment, and ease of method transfer between HPLC and UHPLC. The authors describe a number of potential pitfalls that must be taken into consideration and avoided if accurate and reliable translations are to be achieved. These pitfalls include differences in LC instrumentation dwell volumes, which can lead to coelution of peaks and even reversal of elution order. The difference between the original and new VD/VM ratios must be accounted for: In the case of the new methodology having a larger VD/VM, a delayed injection must be used, or if it has a smaller VD/VM, an isocratic hold must be inserted before commencing the gradient. Errors in translated gradient times of up ~30% can be encountered unless experimentally determined VM values are used. This may affect both retention and selectivity. Hence, the authors strongly recommend that chromatographers practically determine their VM values. The effect of instrument differences between HPLC and UHPLC models have been highlighted as possible sources of translation error; these include differences in column thermostat design, the effect of extracolumn band broadening, and the influence of differing injector design. In addition, the effect of only moderately elevated pressures on selectivity has been demonstrated, coupled with the effect of increased heat of friction associated with smaller particles and the influence of increased efficiency on peak asymmetry has been described.
A free translation tool is available to assist the successful translation between HPLC and UHPLC methods (21). The tool is based on the principles described in this article and permits scaling of gradient times, flow rates, and injection volume as well as accounting for differences in VD/VM ratios between LC systems. A more comprehensive translation tool also will be available (22).
Patrik Petersson is a principal scientist with Novo Nordisk A/S in Måløv, Denmark. Melvin R. Euerby is a professor at the Strathclyde Institute of Pharmacy and Biomedical Sciences in the University of Strathclyde in Glasgow, UK and the head of research and development and training at Hichrom Ltd. in Berkshire, UK. Matthew A. James is a research analyst with Hichrom Ltd.
Direct correspondence to: ppso@novonordisk.com
References
(1) J.W. Thompson, J.S. Mellors, J.W. Eschelbach, and J.W. Jorgenson, LCGC North Am. 24 (s4) , 16–21 (2006).
(2) J.J. Kirkland, F.A. Truszkowski, C.H. Dilks Jr., and G.S. Engel, J. Chromatogr A 890, 3–13 (2000).
(3) J.J. DeStefano, T.J. Langlois, and J.J. Kirkland, J. Chromatogr. Sci. 46, 254–260 (2008).
(4) M. Swartz, LabPlus International 18, 6–9 (2004).
(5) J.R. Mazzeo, U.D. Neue, M. Kele, and R.S. Plumb, Anal. Chem. 77, 460A–467A (2005).
(6) Strategic Directions International (SDi) Tactical Sales and Marketing Report (HPLC: End Users Influencing Development for Advanced HPLC Systems, Columns and Chemistry). (2013).
(7) D. Guillarme, D.T.T. Nguyen, S. Rudaz, and J.L. Veuthey, Eur. J. Pharm. Biopharm. 66, 475–482 (2007).
(8) D. Guillarme, D.T.T. Nguyen, S. Rudaz, and J.L. Veuthey, Eur. J. Pharm. Biopharm 68, 430–440 (2008).
(9) P. Petersson, "Implementation of U(H)PLC within a Global Pharmaceutical Company - A New Way of Working, Advances in High Resolution and High Speed Separations," Chromatographic Soc. Alderley Park, 2010.
(10) R.E. Majors, LCGC North Am. 29 (6), 476–484 (2011).
(11) J.W. Dolan, LCGC North Am. 32 (2), 98–102 (2014).
(12) H. Engelhardt and H. Elgass, J. Chromarogr. A 158, 249–259 (1978).
(13) P. Janderra, J. Chromatogr. A 485, 113–141 (1989).
(14) J.W. Dolan and L. Snyder, J. Chromatogr. A 799, 21–34 (1998).
(15) J.W. Dolan, LCGC North Am. 32 (3), 188–193 (2014).
(16) A.P. Schellinger and P.W. Carr, J. Chromatogr. A 1077, 110–119 (2005).
(17) P. Janderra, J. Chromatogr. A 1126, 195–218 (2006).
(18) A.Clarke, J. Nightingale, P. Mukherjee, and P. Petersson, Chromatography Today, May/June, 4–9 (2010).
(19) www.unige.ch/sciences/pharm/fanal/lcap/telechargement.htm.
(20) P. Petersson and M.R. Euerby, J. Sep. Sci. 30, 2012–2024 (2007).
(21) www.acdlabs.com/translatemymethod.
(22) www.acdlabs.com/products/spectrus/workbooks/chrom/.
(23) European Pharmacopoeia 7th Edition, 2.2.46, "Chromatographic separation techniques," European Pharmacopoeia (European Directorate for the Quality of Medicines, Strasbourg, France, 2011).
(24) A.G. Huesgen, Agilent Technologies, www.agilent.com, Publication Number 5990-5062EN.
(25) P. Petersson and M.R. Euerby, personal communication; unpublished data; March 16, 2014.
(26) A. de Villiers, H. Lauer, R. Szucs, S. Goodall, and P. Sandra, J. Chromatogr. A 1113, 84–91 (2006).
(27) K. Kaczmarski, F. Gritti, and G. Guiochon, J. Chromatogr. A 1177, 92–104 (2008).
(28) Y. Zhang, X. Wang, P. Mukherjee, and P. Petersson, J. Chromatogr. A 1216, 4597–4605 (2009).
(29) F. Gritti and G. Guiochon, Anal. Chem. 80, 5009–5020 (2008).
(30) M.M. Fallas, U.D. Neue, M.R. Hadley, and D.V. McCalley, J. Chromatogr. A 1209, 195–205 (2008).
(31) M.M. Fallas, U.D. Neue, M.R. Hadley, and D.V. McCalley, J. Chromatogr. A 1217, 276–284 (2010).
(32) F. Gritti and G. Guiochon, Anal. Chem. 81, 2723–2736 (2009).
(33) P. Szabelski, A. Cavazzini, K. Kaczmarski, X. Liu, J. Van Horn, and G. Guiochon, J. Chromatogr. A 950, 41–53 (2002).
(34) M.R. Euerby, M. James, and P. Petersson, J. Chromatogr. A, 1228, 165–174 (2012).
(35) S. Fekete, J.L. Veuthey, D.V. McCalley, and D. Guillarme, J. Chromatogr. A 1270, 127–138 (2012).
(36) M.R. Euerby, M.A. James, and P. Petersson, Anal. Bioanal. Chem. 405, 5557 (2013).
(37) N. Wu, A.C. Bradley, C.J. Welch, and L. Zhang, J. Sep. Sci. 35, 2018–2025 (2012).
(38) A.J. Alexander, T.J. Waeghe, K.W. Himes, F.P. Tomasella, and T.F. Hooker, J. Chromatogr. A 1218, 5456–5469 (2011).
(39) P.Petersson, P. Forssen, L Edstöm, F. Samie, S. Tatterton, A. Clarke, and T. Fornstedt, J. Chromatogr A 1218, 6914–6921 (2011).
(40) United States Pharmacopeia General Chapter <621> "Chromatography" (United States Pharmacopeial Convention, Rockville, MD).
(41) U.D. Neue, D. McCabe, V. Ramesh, H. Pappa, and J. DeMuth, Pharmacopeial Forum 35, 1622–1626 (2009).
(42) International Conference on Harmonisation, ICH Q2(R1), Validation of Analytical Procedures: Text and Methodology (ICH, Geneva, Switzerland, 2005).
(43) U.D. Neue, HPLC Columns: Theory, Technology and Practice (Wiley-ICH, New York, 1997), pp. 14.
(44) Japanese Pharmacopoeia 16th Edition, section 2.01 "Liquid Chromatography" (JP XVI 2.01, Pharmaceuticals and Medical Devices Agency, Shin-Kasumigaseki Building, 3-3-2 Kasumigaseki, Chiyoda-ku, Tokyo 100-0013 Japan, 2011).





















Silvia Radenkovic on Her Research and Passion for Scientific Collaboration
April 3rd 2025Radenkovic is a PhD candidate at KU Leuven and a member of FeMS. Her research focuses on inborn metabolic disorders (IMD), like congenital disorders of glycosylation (CDG), omics techniques such as tracer metabolomics, and different disease models.




Evaluating Natural Preservatives for Meat Products with Gas and Liquid Chromatography
April 1st 2025A study in Food Science & Nutrition evaluated the antioxidant and preservative effects of Epilobium angustifolium extract on beef burgers, finding that the extract influenced physicochemical properties, color stability, and lipid oxidation, with higher concentrations showing a prooxidant effect.

