Towards a Universal Detector for Small Molecule Analysis: Direct-EI in LC–MS
LCGC Asia Pacific
Direct-Electron Ionization is a useful tool in LC–MS. This article discusses the potential of this technique as a universal detector for small molecule analysis and how to deal with matrix effects.
Coupling liquid chromatography (LC) and mass spectrometry (MS) is always a 'work in progress' with the final goal of providing the most sophisticated detector capable of identifying and quantifying organic and inorganic compounds at trace level. Well-established LC–MS interfaces and several novel ones are constantly being developed and different operating strategies have been adopted to overcome the apparent difficulties of coupling the two techniques.
LC–MS is complex because LC operates with a highpressure liquid phase, while the MS analyser must operate under high-vacuum gas phase conditions and this appears, in principle, an absolute limitation. The LC mobile phase has a variable composition and high-resolution separations often rely on the use of modifiers that are not always welcome in the MS instrumentation. Physicochemical properties of the analytes can vary dramatically and this influences instrument response, or requires different settings in the interface and in the mass spectrometer.



Advances in Ionization
Despite these limitations, the use of LC–MS has grown in many areas of research due to technical advances in both techniques.
The key of this success is represented by the development of the atmospheric pressure ionization (API) interfaces, among which electrospray ionization (ESI) is the most widely used for medium to highly polar compounds because of its high sensitivity and wide molecular weight (MW) range. Interfacing LC and ESI has produced remarkable advances in biochemical analysis, allowing the mass determination of highmolecularweight, non-volatile biological molecules directly from the liquid phase. This approach is now an invaluable tool in proteomics and has opened many doors in this and other areas of research, including pharmaceutical analysis, environmental analysis, food analysis, bioanalysis and metabolomics.
Atmospheric pressure chemical ionization (APCI) comes into play for small–medium MW and less polar analytes, while the more recently developed atmospheric pressure photo ionization (APPI) and atmospheric pressure laser ionization (APLI) are used for non-polar compounds that are poorly ionized by the other two techniques. However, the ionization of non-polar compounds is still limited in many cases. The ionization is 'soft' in all cases, it takes place through lowenergy chemical processes that lead to a protonated or deprotonated molecular ion (with or without adducts), with a scarce or absent fragmentation. Therefore, only a partial information is obtained and MS–MS, MSn , or high resolution accurate mass MS are required for analyte characterization. The attempt to create reference MS–MS library spectra is a difficult task, still to be fulfilled, because of the number of factors that influence the CID (collision ion dissociation) process into an MS–MS instrumentation.
Many authors report that quantitative analysis in ESI and, to a lesser extent, in APCI, is affected by the presence of interferences from the matrix that induce ion suppression or enhancement.1–5 Matrix effects (ME) are responsible for unreliable quantitative data, even within a series of replicates, scarce reproducibility, linearity and accuracy of the overall method. ME are highly dependent on the sample nature and the unpredictable response that they induce can be detected, even among different lots of the same sample, or with the same method. A possible explanation relies on the assumption that the analyte and the co-eluted matrix can compete in the liquid phase for the available charges and for the access to the droplet surface for gas-phase emission.6,7 During method development each step should be optimized to reduce ME. Sample clean-up procedures, more efficient chromatographic separations and novel mass spectrometric approaches can play a significant role, together with appropriate data analysis. Nevertheless, soft ionization techniques are not very well suited for the analysis of true unknown compounds, such as in gas chromatography–mass spectrometry (GC–MS), in which 70 eV electron ionization (EI) allows the collection of library spectra for sample identification. Therefore, the demand for new LC–MS instrumentation is strongly increasing in many applications.
Electron Ionization
A niche of LC–MS instrument development involves the use of electron ionization. From a historical standpoint, it is worth mentioning that EI pioneered the LC–MS development but, despite an initial success, mainly offered by the particle beam (PB) interface,8 it was rapidly outpaced by the development of the soft ionization techniques. They opened the way to the analysis of molecules of biological interest, attracting most researchers' attention. Consequently, the efforts to develop a more efficient EI-based interface were cast off, although a considerable number of small–medium molecular weight substances can provide very good EI spectra. The operating principle is simple and it is based on the gas-phase ionization of the sample, disregarding its chemical background, as long as it can withstand the typical EI source conditions (high vacuum and temperature). The absence of ion–molecule or ion–ion reactions reduces the effect of the mobile phase in the ionization process reducing ME, avoiding any post-column adjustments.
Similarly in GC–MS, the spectrum obtained is very informative; its high reproducibility allows an easy comparison with the spectra from commercially available electronic resources (such as NIST or Wiley). It can also benefit the algorithms developed by the National Institute of Standards and Technology (NIST) and Automated MS Deconvolution and Identification System (AMDIS), which extract the analytes' mass spectra in complex chromatographic mixtures in the presence of several overlapping peaks. The identification is automatic, legally defensible and provides structural information that can help to identify unknown compounds.9
However, we cannot understate the fact that the operating conditions of EI and LC are quite dissimilar and this might explain some of the limitations observed in the past. Poor sensitivity, signal instability, reduced linearity and disappointing optimization were everyday frustrations, especially when working in reverse-phase conditions. Various authors attempted to efficiently generate EI spectra from a liquid effluent. Kientz et al.10 were able to generate EI spectra based on an eluentjet formation by means of inductive heating of the micro-LC effluent and momentum separation in a jet separator; Dijkstra et al.11 improved that technique with a particular emphasis on its use under chemical ionization (CI)–MS conditions. A considerable effort in the exploitation of EI in LC–MS has been done by Amirav et al., who explored the potential of supersonic molecular beams (SMB-MS) to produce intense molecular ions in EI spectra from an LC effluent. The authors called this approach Cold EI to emphasize that the SBM serves as a medium for EI of vibrationally cold molecules.12,13 A spray of liquid is formed at high pressure, then the sample particles are vaporized prior to sample expansion, as isolated molecules, from a supersonic nozzle.
A supersonic molecular beam, containing vibrationally cold sample molecules, is then collimated and proceed axially through an EI ion-source to obtain Cold EI mass. The spectra provide an enhanced molecular ion, but also library-searchable fragments for improved confidence level in the identification. Our research group has been working for more than a decade and through several designs to develop an up-to-date LC–MS based on EI. Keeping in mind the operating principles of PB, we significantly improved sensitivity for any mobile-phase composition by drastically reducing the mobile-phase intake.14 This first approach could be considered as a miniaturization of the PB interface, therefore, we called it microparticle beam and it was followed by its evolution, called capillary EI (CapEI, commercialized by Waters, Milford, Massachusetts, USA, in 1999). Both devices used microscale flow rates (1–5 µL/min) and they were successfully tested in the analysis of several classes of compounds under different reverse-phase HPLC conditions.15–24 In particular, for the first time, a significantly improved tolerance for nonvolatile buffers was observed in mass spectrometry.25,26 Taking advantage of the extremely low flow rate typical of the nano-HPLC columns, we conceived a completely new interface, particularly simple in the design. The effluent from the column is directly introduced into the ion source at nano-scale flow rates (< 500 nL/min) with the advantage to further reduce the adverse effects of mobilephase vapours. We called it Direct-EI because the nano-column is directly coupled to the MS, with the same simplicity of a capillary GC column, without any intermediate apparatus.
The Direct-EI Interface
One of the most challenging efforts in LC–MS is to eliminate the solvent vapours before they enter the high vacuum region. In ESI interface, despite the fact that the liquid phase plays a fundamental role in the ionization process, it must be removed prior to entering the analyser, because its instantaneous vaporization is sufficient to raise the pressure to inacceptable values. This operation should be performed very carefully to avoid sample losses and interferences with the ionization process, with the aim to generate a solvent-free ion beam focused into the high vacuum analyser region. In EI this issue must be addressed at an early stage, in fact the typical ion source conditions are high vacuum (approx. 10–5 torr) and high temperature, therefore, only a very limited liquid intake should be allowed into the ion source. The development of nanoscale LC columns has allowed the separation of complex mixtures at nanolitres per minute flow rate. This is a sufficiently low flow rate to be introduced into a modified EI ion source, without breaking the vacuum and compromising the correct functioning. Keeping in mind these considerations, we developed the Direct-EI interface, in which the entire interfacing process takes place in the small volume of the ion source, with a simplicity comparable to a GC–MS system. The liquid phase enters the interface via a capillary connecting tubing.

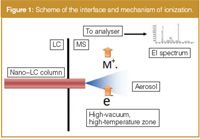
Figure 1
As illustrated in Figure 1, an aerosol is formed in high vacuum conditions, followed by a quick droplet desolvation and final vaporization of the solute, upon contact on a target surface prior to the ionization. The tip of the nebulizer is extended into the ion source so that the spray expansion is completely contained inside the volume. The eluate must reach the ion source as liquid phase and to prevent a premature in-tube solvent evaporation the nebulizer and the connecting tubing must be thermally insulated from the source heat. This is very important to obtain a good quality aerosol and to avoid solute precipitation that might clog the tip. The solute conversion to the gas phase is promoted by the source heat.
The temperature is kept between 200 °C and 350 °C and has a double function: to compensate for the latent heat of vaporization during the droplet desolvation and to convert the solute into the gas phase upon contact on the source surface. The transition from liquid to aerosol is very rapid and the mix of vapours is homogeneously distributed in the ion volume. The flow rate introduced should not exceed 0.75 µL/min for most reverse mobile phase compositions, mainly imposed by the column backpressure. It normally generates less than a mL/min of vapour, which is within the pumping capacity of most MS systems, and it is roughly the same of the carrier gas (helium) introduced by a conventional GC capillary column. At this flow rate we did not report any adverse effects on the MS hardware (filaments, lenses, insulations, etc.). Conversely, the lowest flow rate capable of generating a fine and homogeneous spray is approximately 100 nL/min. The design of the interface allows the fast removal of the solvent, improving the signal-to-noise (S/N) ratio and the sensitivity, and eliminating chemical ionization interferences. Solvent signals can be detected in the low mass range of the mass spectrum recorded, but they can easily be subtracted, preserving the intrinsic quality of the EI spectrum.
Another advantage offered by the nano-columns is that the reduced flow rate allows the introduction of a steady stream of non-volatile buffers, as mobile-phase modifiers, into the ion source. Our previous EI based interfaces, and now the Direct-EI, demonstrate that the combination of reduced flow rate and EI enhances the tolerance towards chemically modified mobile phases.25,26 Many chromatographic separations benefit from the presence of non-volatile buffers in the mobile phase. In ESI, non-volatile species cause salt depositions on the metal surfaces and could completely block ion transmission. In addition, if the anion and the cation pair too strongly with the analyte, then the analyte ions might be prevented from carrying the excess charge on the droplet surface; as a result, the ESI response may be very low. For this reason volatile buffers are used instead. In EI the analyte is ionized in the gas-phase independently and the signal is not influenced by the presence of pre-formed ions in the form of salts or strong acids and bases. Routine cleaning is enough to get rid of the very limited salt deposition into the ion source. For the same reasons a wider selection of solvents can be used as well.
We already mentioned that quantification procedures based on ESI and APCI can be severely affected by ME. In Direct-EI, as opposed to API techniques, the gas-phase ionization process is a physical-based odd electrons one and involves a high-energy content. Fragmentation is spontaneous, pathways are highly reproducible and independent from the type of instrument. Co-eluted components are ionized independently from each other, so that signal intensity is invariably related with the concentration of each analyte. In ESI the ionization takes place in the liquid phase, so that any co-eluted substance from the matrix can interfere with the available charges inducing either ion suppression or enhancement of the signal.
This interface was tested on an Agilent 5475 MSD (Agilent Technologies Inc., Santa Clara, California, USA), and compared to our previous designs, it shows the following improvements: (a) LODs to the low-picogramme level for most substances in SIM; (b) higher spectral quality with full spectrum searching capability using electronic libraries; (c) no chemical ionization (CI) signals in the mass spectrum with highly accurate reproduction of isotope ion clusters; (d) full compatibility with nonvolatile buffers and other salts in the mobile phase; (e) absence of ME in quantitative applications; (f) extended range of linear response; (g) good intra- and interday signal stability.
Fields of Applications
Direct-EI offers a valid and reliable alternative to other LC–MS interfaces for a large number of small–medium molecular weight molecules, with the only condition to withstand a gas phase ionization. The diagram in Figure 2 demonstrates how 101 compounds, spanning from n-alkanes to sugars, with very different polarity and molecular weight can be successfully ionized with Direct-EI. This interface was tested in many fields of applications (environmental, pharmaceutical and biological) under different analytical conditions, at trace-level concentrations and with various matrices.27 In many aspects, Direct-EI can bridge the gap between GC–MS and LC–MS because we found that many compounds that are chemically sensitive or difficult to vaporize by GC–MS can take advantage of the nano-flow nebulization and give stable and intense signals (i.e., fatty acids, phenoxyacid pesticides). Other analytes that give a poor response, or do not ionize at all with ESI can be successfully analysed with Direct-EI (i.e., organochlorine pesticides, diethylene glycol).

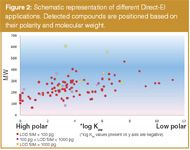
Figure 2
Multiresidue analysis of substances with different polarities can be highly frustrating. In fact, the protocols for low-polar, thermostabile analytes determination are traditionally based on gas chromatography (GC), mainly coupled to detection techniques such as electron capture detection and mass spectrometry.28–31 Conversely, LC–MS represents the method of choice for high-polar, poorly volatile compounds,31,32 but falls short when low polar compounds are present as well. Twelve OCPs together with seven phenoxyacid pesticides in water samples, with different physicochemical properties, were selected and analysed in a single step with Direct-EI-MS, after off-line SPE sample clean-up. Thanks to the gas-phase ionization, no ME were reported, regardless of the complexity of the original matrix. In Figure 3 another application of environmental interest regarded the analysis of organophosphorus pesticides and endocrine disrupting compounds (EDC) in water samples.


Figure 3
A similar example regards the analysis of fragrances and other perfumery compounds. Figure 4 shows a scan profile of 36 substances at a concentration of 1 mg/mL, almost completely separated by the column. Some of the analytes were not GC amenable, but gave an uneven response with ESI. For all of them it was possible to record interpretable EI mass spectra as well as for three interferences present in the original mixture.


Figure 4
To further test the performance of Direct-EI, and to assess its behaviour in terms of ME, we selected ibuprofen and phenacetin as model compounds and human plasma as complex matrix. Plasma samples were cleaned-up with two different procedures: liquid–liquid extraction and solid-phase extraction and ME was evaluated using the post-extraction addition method. The blank extracts were spiked with a given amount of a standard of the analytes and analysed in parallel with LC–ESI-MS–MS and LC-Direct-EI-MS. The effect of matrix composition on the response of the two interfaces for the target analytes is summarized in Table 1. In ESI-MS–MS analysis the presence of the matrix induced a consistent signal suppression for ibuprofen, whereas we observed signal enhancement for phenacetin. This trend was reported in all extracts with a drastic effect on both method precision and accuracy, regardless of the clean-up procedure used for sample preparation. On the contrary, the response with Direct-EI was not influenced by the presence of the matrix, under any circumstance. These results demonstrate that the method based on LC–ESI-MS–MS cannot be validated in the present form; therefore, strategies to address ME are mandatory.


Table 1
Another application involves the profiling of free fatty acids (FFA) in plasma samples.37 Preliminary results with LC-Direct-EI-MS analysis have shown several advantages over the current protocols that entail GC–MS and LC–API-MS methods. First of all, the sample preparation is faster because it does not require the compounds' derivatization. The response of the target compounds is not influenced by ME, and reproducible, high-quality library-matchable spectra are obtained. On the other hand, although sensitivity is satisfactory for the quantification in human plasma, it is not as high as that of GC–MS and LC–API-MS. Our future efforts will be dedicated to increasing sensitivity.
Conclusions
The Direct-EI represents a valid alternative in LC–MS for the analysis of small–medium molecular weight compounds in a wide range of polarities, with a sufficient thermal stability. A large number of applications can benefit from this approach that fills the gap between GC–MS and API-based LC–MS. Our experiments demonstrate that Direct-EI is a robust and stable platform, where a "direct", simple approach combines with high operational flexibility and a large extent of information with an affordable instrumentation. Its performance is scarcely influenced by compound structure and mobile phase composition. In addition, the reduced flow rate allows the introduction of non-volatile modifiers and to limit ME thanks to the gas-phase ionization. Future efforts will be addressed to increase sensitivity, in which Direct-EI is still less competitive with ESI in terms of very low detection limits for specific analytes.
Acknowledgements
The authors would like to thank Agilent Technologies for their support with this research.
Pierangela Palma is an assistant professor at the University of Urbino, where she teaches Analytical Chemistry. Her research focuses on new advances in micro- and nano-HPLC columns, and on the development of LC–MS techniques in wide range of applications.
Giorgio Famiglini is assistant professor of analytical chemistry at the University of Urbino. His principal field of interest is LC–MS coupling, but also has experience in method development in complex biological and environmental matrices.
Helga Trufelli graduated at the University of Urbino in 2000, and she earned her PhD in Chemical and Pharmaceutical Sciences at the same University in 2004. Since 2004 she has been involved in the LC-Direct-EI-MS project as a postdoctoral associate.
Achille Cappiello is currently a professor of Analytical Chemistry at the University of Urbino. He is the director of several projects dealing with instrument development and design on LC–MS with some of the major instrument manufacturers and actively collaborates with the pharmaceutical industry with innovative LC–MS applications.
References
1. B.K. Matuszewsky, M.L. Constanzer and C.M. Chavez-Eng., Anal. Chem., 75, 3019–3030 (2003).
2. J.P. Antignac et al., Anal. Chim. Acta, 529, 129–136 (2005).
3. P.J. Taylor, Clin. Biochem., 38, 328–334 (2005).
4. W.M.A Niessen, P. Manini and R. Andreoli, Mass Spectrom. Rev., 25, 881–899 (2006).
5. L. Tonidandel and R. Seraglia, Journal of Chromatography Library, 72, A. Cappiello, Ed., 193–210 (Elsevier, Amsterdam, The Netherlands 2007).
6. N.B. Cech and C.G. Enke, Anal. Chem., 72, 2717–2723 (2000).
7. R. King et al., J. Am. Soc. Mass Spectrom., 11, 942 (2000).
8. R.C. Willoughby and R.F. Browner, Anal. Chem., 56, 2625–2632 (1984).
9. F.W. McLafferty, "Interpretation of Mass Spectra" University Science Books, Mill Valley, California, USA (1980).
10. C.E. Kientz et al., Anal. Chem., 68, 675–681 (1996).
11. R. Dijkstra et al., Rapid Commun. Mass Spectrom., 12, 5–10 (1998).
12. A. Amirav and O. Granot, J. Am. Soc. Mass. Spectrom., 11, 587–591 (2000).
13. A. Amirav, Int. J. Mass. Spectrom., 244, 15–28 (2005).
14. A. Cappiello and F. Bruner, Anal. Chem., 65, 1281–1287 (1993).
15. A. Cappiello, Mass Spectrom. Rev., 15, 283–296 (1996).
16. A. Cappiello, G. Famiglini and F. Bruner, Anal. Chem., 66(9), 1416–1423 (1994).
17. A. Cappiello and G. Famiglini, Anal. Chem., 67(2), 412–419 (1995).
18. A. Cappiello et al., J. Am. Soc. Mass Spectrom., 6, 132–139 (1995).
19. A. Cappiello, G. Famiglini and B. Tirillini, Chromatographia, 40(7–8), 411–416 (1995).
20. A. Cappiello et al., Environ. Sci. Technol., 29(9), 2295–2300 (1995).
21. A. Cappiello et al., J. Am. Soc. Mass Spectrom., 7, 753–758 (1996).
22. A. Cappiello and G. Famiglini, J. Am. Soc. Mass Spectrom., 9, 993–1001 (1998).
23. A. Cappiello et al., J. Chromatogr. A, 855, 515–527 (1999).
24. A. Cappiello et al., Anal. Chem., 72, 3841–3846, (2000).
25. A. Cappiello et al., Anal. Chem., 69, 5136–5141 (1997).
26. A. Cappiello et al., Environ. Sci. Technol., 33, 3905–3910 (1999).
27. A. Cappiello et al., Mass Spectrometry Reviews, 24, 978–989 (2005).
28. F. Mangani, M. Maione and P. Palma, Handbook of Water Analysis, L.M.L. Nollet, Ed., 517–536 (Marcel Dekker, New York, 2000).
29. M. Barriada-Pereira et al., Chemosphere, 1571–1578 (2005).
30. S. Yenisoy-Karakasü, Anal. Chim. Acta, 571, 298–307 (2006).
31. M. Guardia-Rubio et al., Microchem. J., 85, 257–264 (2007).
32. T. Reemtsma, J. Chromatogr. A, 1000, 477–501 (2003).
33. M. Kuster, M. Lopez de Alda and D. Barceló, Mass Spectrom. Rev., 25, 900–916 (2006).
34. G. Famiglini et al., Anal. Chem., 80(9), 3445–3449 (2008).
35. A. Cappiello et al., Anal. Chem., 74, 3547–3554 (2002).
36. G. Famiglini et al., Anal. Chem., 77, 7654-7661 (2005).
37. P. Palma et al., 34th International Symposium on High-Performance Liquid Phase Separations and Related Techniques International Congress (HPLC 2009), Dresden, Germany 28 June–2 July (2009).




















New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.




