Rapid Adeno-Associated Virus Genome Integrity Analysis by Capillary Gel Electrophoresis
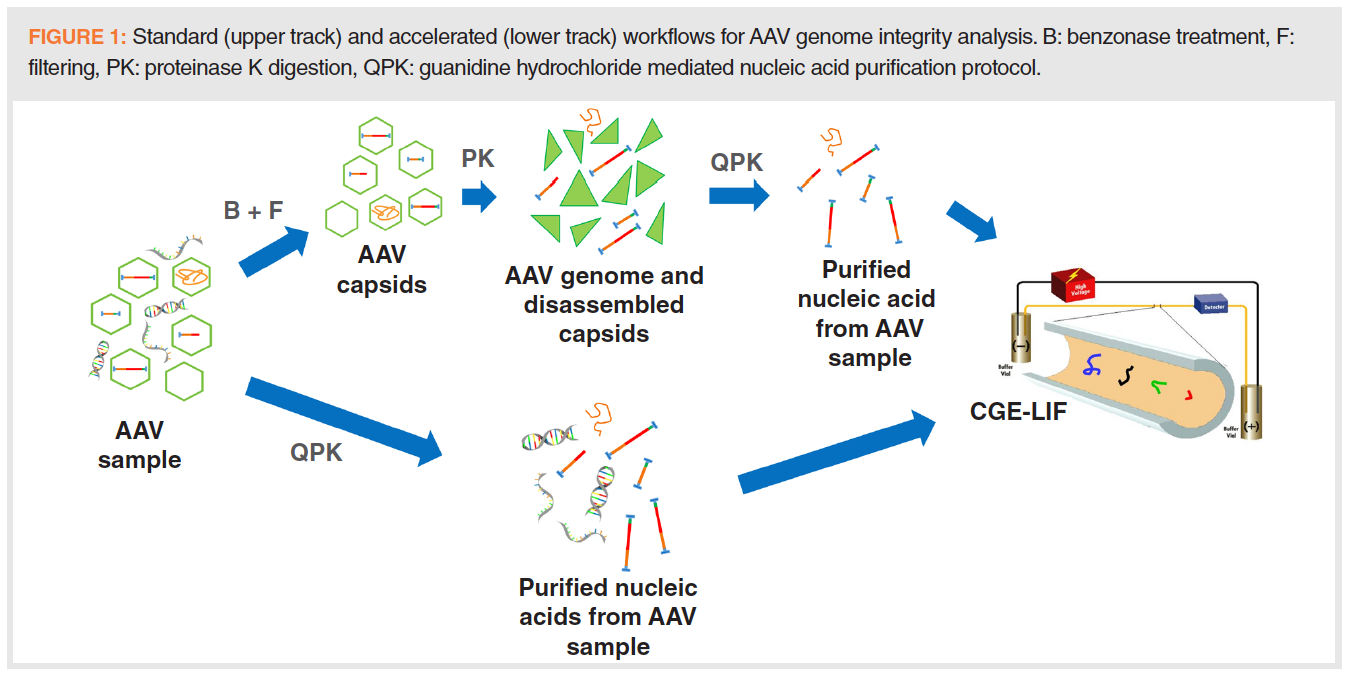
In this paper, two adeno-associated virus (AAV) genome integrity analysis workflows are introduced; a standard sample preparation protocol and an accelerated procedure, both utilizing capillary gel electrophoresis with laser induced fluorescent detection (CGE–LIF) to analyze the released nucleic acids.
Adeno-associated virus (AAV) is a propitious gene therapy vector to be translated to the clinical practice and pursued accordingly by the biopharmaceutical industry. However, genome integrity and nucleic acid impurities in the final product may represent a problem with the therapeutic use. In this paper, two AAV genome integrity analysis workflows are introduced. The standard sample preparation protocol (~2.5 h) included benzonase treatment to degrade any nucleic acids outside of the AAV capsid, which were then filtered out. The residual AAV capsids were digested by proteinase K, followed by a DNA purification process involving guanidine–hydrochloride. The accelerated protocol (~15 min) utilized the DNA purification step only. In both workflows, the released DNA was analyzed by capillary gel electrophoresis with a laser induced fluorescent detection (CGE–LIF) in less than 15 min with ≤ 0.33% RSD on migration time and ≤ 2.06% RSD on peak area reproducibility.
Adeno‑associated viruses (AAVs) are non‑enveloped icosahedral protein shell capsids of ~20 nm in diameter, containing a single‑stranded DNA genome of about 4.7 kb in length (1). AAV vectors feature no pathogenicity, low immunogenicity, wide tropism for multiple cell types, and persistent transgene expression for both dividing and quiescent cells (2). Therefore, AAV represents a strong translational option to treat a plethora of diseases (3). The genome of recombinant AAV comprises two inverted terminal repeats required for multiplication and encapsidation, a promoter region to regulate transgene expression, the transgene encoding the therapeutic protein, and a polyadenylation signal for RNA stabilization (4). Since there are no Rep and Cap gene sections in the transgene‑containing AAV genome, it is replication deficient.
One of the main challenges during AAV manufacturing is the characterization and identification of product‑related impurities at the genome integrity level as its quality inside the viral vector may affect infectivity, efficacy, and safety (5). The most important of such quality issues are instances when the transgene‑containing genome is not present or only partially present (truncated); or the capsid contains fragments of host cell genome or plasmid DNA. Therefore, genome integrity and purity analysis is crucial, requiring robust, reliable, and high resolution methods. Techniques currently used for such analysis include denaturing agarose gel electrophoresis, Southern blot tests, quantitative polymerase chain reaction (qPCR), size‑exclusion high‑performance liquid chromatography (SEC–HPLC), and next generation sequencing (6). Denaturing agarose gel electrophoresis is a low cost and easy‑to‑use method, but time consuming with limited precision (7). Southern blotting is another conventional technique that can confirm the presence of the target gene with correct size determination, however, suffers from low precision and cannot detect impurities (8). Real time polymerase chain reaction (PCR) can readily quantitate the target AAV genome sequence, but does not provide size determination and cannot detect fragments that do not contain the target sequence (9,10). SEC–HPLC features good sensitivity, reproducibility, and quantification of the intact genome and other DNA fragments in the capsid, nevertheless lacks adequate size assessment (11). Finally, next generation sequencing can determine the sequence of the AAV genome along with any contaminants, although cannot be easily adopted as a routine quality control test in the biopharmaceutical industry (12).
Capillary gel electrophoresis (CGE) is a well-established automated bioanalytical method, especially applicable at the nucleic acid separation field, where it was extensively used for the initial sequencing of the human genome (13). Equipped with laser induced fluorescent (LIF) detector, a limit of detection (LOD) down to 10-11 M can be routinely obtained (14). In this paper, a standard and an accelerated sample preparation workflow (Figure 1) are introduced in conjunction with CGE–LIF separation for AAV genome integrity and purity analysis.


Materials and Methods
Materials: The SYBR Green II RNA gel stain (10 000 × in DMSO) and the 10× DNase I buffer were obtained from Thermo-Fisher Scientific (Waltham, Massachusetts, USA). Polyvinylpyrrolidone (PVP, MW 1.3 MDa), benzonase, EDTA, Transcript RNA markers (0.2–10 kb), 10× Tris Borate EDTA (TBE) buffer, and the Amicon Ultra-0.5 centrifugal filter units (MW cutoff: 100 kDa), and all other chemicals were from Millipore Sigma (St. Louis, Missouri, USA). The QIAquick PCR purification Kit and Proteinase K were from Qiagen (Germantown, Maryland, USA). Packaged AAV8 serotype (titer: ~1013 GC/mL) and its formulation buffer (1× PBS with 0.001% Pluronic F68) were from Vigene Biosciences (Rockville, Maryland, USA). The AAV2 and AAV5 serotype samples were from SignaGen (Rockville, Maryland, USA).
AAV Genome Preparation: The nucleic acid impurities in the AAV samples were digested in 20 µL reaction volume containing 10 µL of sample, 7 µL of formulation buffer, 2 µL of 10× DNase I buffer, and 1 µL tenfold diluted benzonase in 1× DNase I buffer of 10 mM Tris‑HCl (pH 7.5), 2.5 mM MgCl2, and 0.1 mM CaCl2. All digestions were performed for 30 min at 37 °C and terminated by the addition of 2 µL of 50 mM EDTA and 278 µL of AAV formulation buffer. Benzonase and any degraded nucleic acids were removed by centrifugation for 2 min at 14 000 × g through an Amicon centrifugal filter (100 kDa cutoff). The residue was collected by inverting the filter unit, spun for 2 min at 1000 × g and diluted with the AAV formulation buffer (95 µl). AAV capsids were disassembled by the addition of 5 µL of 20 mg/ml Proteinase K and incubated for 2 h at 37 °C. The released AAV genome was purified by the QIAquick PCR Purification Kit including guanidine HCl, and eluted by 30 µL of nuclease free water. The eluted sample was incubated for 2 min at 70 °C and put on ice before CGE–LIF analysis.
Capillary Gel Electrophoresis: The PA 800 Plus Pharmaceutical Analysis System equipped with a solid‑state laser induced fluorescent detector (excitation wavelength 488 nm, emission band pass filter: 520 nm) and the pre-assembled EZ‑CE Capillary Cartridge were from Sciex (Framingham, Massachusetts, USA). The separation gel‑buffer system contained 1% PVP (1.3 MDa), 89 mM Tris – Boric acid (pH 8.3), 2 mM EDTA (1× TBE), 4 M urea, and SYBR Green II (1–25 000 dilution). The samples were injected electrokinetically at 5 kV applied potential for 3–10 s. The separation voltage was 6 kV (200 V/cm) in reversed polarity mode (cathode at the injection side). Data acquisition and analysis were performed using 32 Karat software V10.3.
Results and Discussion
The main goal of this work was to develop rapid, but reliable analytical methods to assess the quality and size of encapsulated AAV genomes. A standard and an accelerated sample preparation protocol were applied and the released DNA molecules were separated by CGE (Figure 1). The standard workflow, shown in the upper track of Figure 1, started with treating the AAV samples with benzonase to digest all nucleic acid contaminants outside of the capsid. This was followed by filtering (100 kDa cutoff) to remove the enzyme and all small size oligonucleotides (~ 4-6 bases long). The next step of the standard workflow was to break down the capsid with proteinase K, followed by purification of the released DNA using a guanidine hydrochloride mediated protocol. The released DNA was then analyzed by CGE using LIF detection for enhanced sensitivity. The lower track in Figure 1 delineates the accelerated method skipping both the benzonase and proteinase K treatments, only utilizing the DNA purification step (involving guanidine hydrochloride) before CGE–LIF analysis.
Evaluation of the Two Workflows
First, the standard workflow (Figure 1, upper track) was applied to an AAV8 sample with the genome size of 4.5 kb and the released nucleic acids were analyzed by CGE–LIF. This sample preparation route was initiated with benzonase digestion. Using this non‑selective endonuclease, all nucleic acid impurities possibly present in the samples outside of the AAV capsid such as DNA and RNA (probably originated from degraded host cells and/or the plasmid vector) were broken down to very short oligonucleotides (~ 4-6 bases). After the digestion step, the reaction mixture was filtered through a 100 kDa cutoff filter to remove the benzonase and the digested small nucleic acid molecules. The filtration residue, containing the AAV capsids, was subject to proteinase K treatment to release its genome content, which was cleaned by a guanidine hydrochloride-mediated DNA purification protocol before CGE–LIF analysis. The upper trace in Figure 2 shows the resulting electropherogram of the extracted AAV8 genome following the standard protocol. Lacking commercially available single stranded DNA standards at the size range of up to 10 kb, approximate sizing was established by using an RNA ladder (0.2–10.0 kb, fragment sizes are given by the upper x-axis in the figures), with the assumption that single stranded RNA species migrate similarly to that of their single stranded DNA counterparts. Based on the size assessment, as a first approximation we considered Peaks 1 and 2 as smaller and Peak 3 as larger size impurities in the 0.5–0.9 and 0.9–1.3 kb ranges, respectively. Peak 4 is the 4.5 kb intact genome of the AAV8 viral vector. The small Peaks 5 and 6 are probably different secondary structure forms of the intact genome. The migration time and peak area reproducibility of the separated components of interest in Figure 2 (trace a, Peaks 1, 2, 3, and 4) were ≤ 0.33% RSD and ≤ 2.06 %RSD, respectively, based on eight consecutive runs.

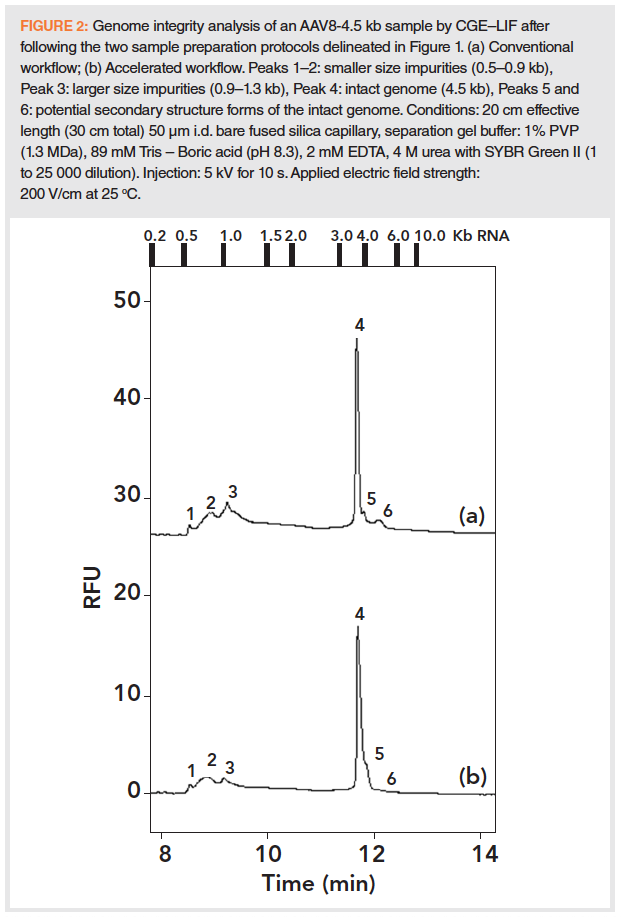
An accelerated and simplified sample preparation approach was also evaluated (lower track in Figure 1) to shorten the time requirement of approximately 2.5 h of the standard workflow. Here, the AAV8‑4.5 kb sample was neither digested by benzonase, nor by Proteinase K, but purified with a spin column using the method described in the experimental section. Apparently, the guanidine hydrochloride content in the purification binding buffer sufficiently opened up the capsids allowing to release its DNA content for downstream CGE–LIF analysis. In this instance, the entire sample preparation and CGE–LIF analysis procedure required less than 30 min. Trace b in Figure 2 shows a very similar electropherogram to trace a, where the standard sample preparation approach was used, but with somewhat decreased signal intensity. Here, Peak 2 is broader than the corresponding peak in trace a, suggesting inclusion of some nucleic acid impurities from outside of the capsid, that this approach does not remove. In trace b, Peak 5 migrated as a tailing shoulder of Peak 4 and Peak 6 was almost not visible. If such slight decrease in sensitivity is acceptable, this accelerated method can be the choice of use during AAV manufacturing as a fast in‑process analytical technology approach, with the understanding that nucleic acids outside of the capsids are co‑measured with the ones inside. Please note that the injection parameters can be optimized in the rapid method to increase detection sensitivity.
Analysis of the Origin of Impurities
As suggested above, the origin of the smaller size impurities in Figure 2 (Peak groups 1 and 2) were considered as host cell or plasmid related fragmented nucleic acid artifacts. To prove this assumption, both full and empty AAV8 samples, the latter containing almost no viral genome, were evaluated with and without benzonase treatment, with the assumption that benzonase digests all nucleic acid impurities outside of the viral capsid. In both instances, the samples were centrifuged through a 100 kDa filter and the residues were subject to proteinase K digestion. All samples were then purified with the guanidine hydrochloride mediated method as described in the Experimental section and analyzed by CGE–LIF. The quantitative results based on the resulting peak areas are compared in Table 1. The non‑benzonase treated (-B), proteinase K digested (+PK) and purified (+gHCl) empty AAV8 vector showed the highest amount of smaller size nucleic acid impurities (peak area: 133247), apparently representing the total amount of impurities in this particular sample. Applying the benzonase treatment to the same empty capsid (+B), followed by filtration and proteinase K digestion (+PK) and purification (+gHCl), on the other hand, significantly reduced the amount of nucleic acid impurities detected (peak area: 59341). Small peaks representing the intact genome were also detected in these two empty capsid samples with peak areas of 802 and 691, respectively. This could be manufacturing related, for instance due to the co‑collection of a small amount of full capsids together with the empty capsids during the preparation of the empty capsid sample after iodixanol density gradient ultracentrifugation.

As a control, full AAV8-4.5 kb samples were processed by the same way as described above with and without benzonase treatment (+B/-B) followed by proteinase K digestion (+PK) and guanidine hydrochloride mediated sample purification (+gHCl). The quantitative results obtained with these two samples are also listed in Table 1. As expected, practically the same amount of intact genome was found after both sample processing steps. Please note that measuring the peak area differences between the full (9914 and 10161) and empty capsid (802 and 691) might offer a titer determination prospective. On the other hand, the low peak area values of the smaller size impurity peaks appeared in the full AAV8 capsid analysis with and without benzonase treatment (+B: 1585 and –B: 2019) indicated apparently high sample purity. Some larger size impurities were also detected with peak areas of 909 and 619, for the benzonase treated and non-treated full capsid samples, respectively, providing sufficient information about the integrity of the AAV genome. Based on these quantitative results, it was considered that the larger size impurities were most likely from inside of the capsids, while the smaller size impurities were present both outside and inside of the capsids.
Application of the Accelerated Protocol
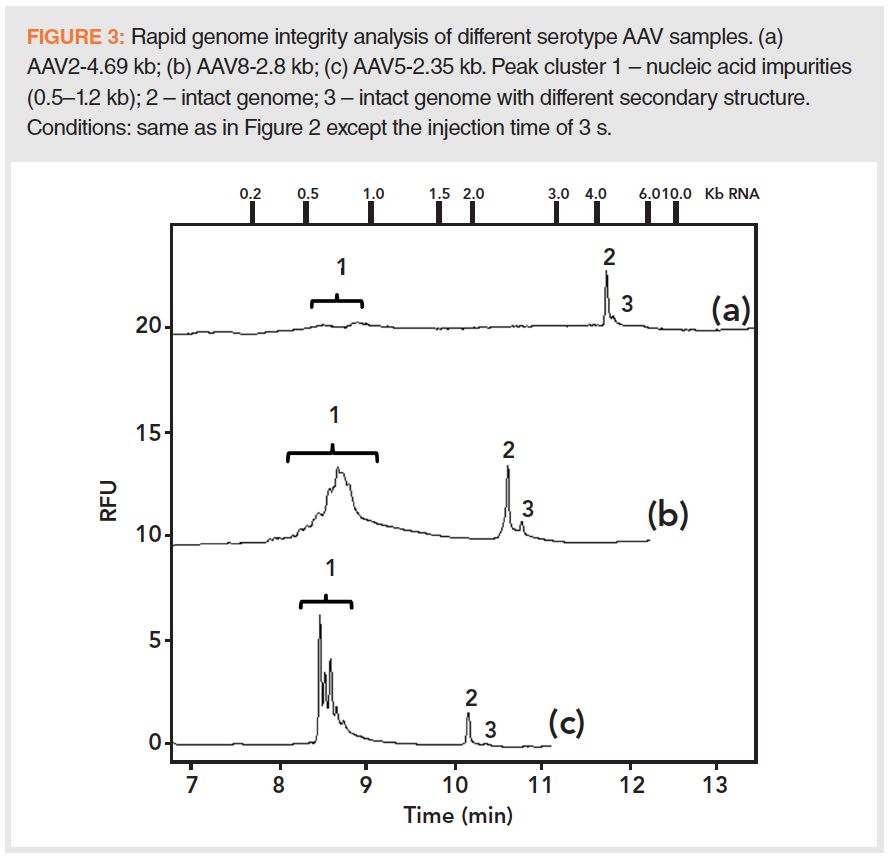
The accelerated sample preparation protocol was applied to the following three samples with different serotypes: AAV2 (4.69 kb genome), AAV8 (2.8 kb genome), and AAV5 (2.35 kb genome). The resulting electropherograms are compared in Figure 3. The AAV2 sample (trace a) was found to be reasonably clean, with a few small peaks (Peak group 1) representing smaller size impurities (0.5–0.9 kb) and Peak 2, the genomic DNA load. Peak 3 is probably the same intact genome but with a different secondary structure, separated by the high resolving power of CGE. The middle trace (b) depicts the CGE–LIF analysis of the released DNA from the AAV8-2.8 kb sample. Here the broad Peak 1 depicts the cluster of all smaller and larger size impurities in the sample ranging approximately from 0.2 to 1.2 kb, while Peak 2 represents the intact 2.8 kb DNA. Similar to trace a, Peak 3 is probably a different secondary structure bearing intact genome. The lower trace (c) shows the electropherogram of the AAV5‑2.35 kb sample. Similar to the other traces, smaller size impurities were also detected here with better resolution and a somewhat narrower size range of around 0.5–0.8 kb. These minor variations were probably due to the actual manufacturing processes applied.

Conclusions
In this paper, a standard and an accelerated sample preparation method were compared for genome integrity analysis of AAVs to assess their size and quality as well as the proportion of the impurities possibly present. The standard 2.5 h workflow provided more in-depth analysis including the benzonase treatment to remove any nucleic acid impurities outside of the capsid simplifying in this way the data interpretation. Genome integrity analysis using the accelerated workflow (skipping both the benzonase and proteinase K digestion steps, but still having the guanidine hydrochloride mediated purification step), on the other hand, took only 30 min including CGE–LIF separation, but did not allow differentiation of nucleic acid impurities outside and inside the capsid. The accelerated workflow was applied to the analysis of various AAV serotypes with different size genome loads. Based on the results, the accelerated method could be considered appropriate for fast in-process quality testing during AAV manufacturing, however, for in‑depth impurity analysis the standard technique is recommended. It is important to note that the analysis methods presented in this paper may have the potential for titer determination upon further fine tuning.
Acknowledgments
The authors greatly appreciate the helpful discussions with Elliott Jones, Sahana Mollah, Fang Wang, Tingting Li, and Zaifang Zhu. This is contribution #174 from the Horváth Csaba Memorial Laboratory of Bioseparation Sciences.
References
- M.F. Naso, B. Tomkowicz, W.L. Perry III, and W.R. Strohl, BioDrugs31, 317–334 (2017).
- K.M. Van Vliet, V. Blouin, N. Brument, M. Agbandje-McKenna, and R.O. Snyder, Methods Mol. Biol.437, 51–91 (2008).
- J.F. Wright, Biomedicines2, 80–97 (2014).
- R. dos Santos Coura and N. Beyer Nardi, Genet. Mol. Biol.31, 1–11 (2008).
- H.C. Verdera, K. Kuranda, and F. Mingozzi, Mol. Ther.: the journal of the American Society of Gene Therapy28, 723–746 (2020).
- J.F. Wright and O. Zelenaia, Methods Mol. Biol.737, 247–278 (2011).
- E. Kohlbrenner and T. Weber, Methods Mol. Biol.1521, 91–107 (2017).
- B. Dong, H. Nakai and W. Xiao, Mol. Ther.: the journal of the American Society of Gene Therapy18, 87–92 (2010).
- M. Andre, S. Reghin, E. Boussard, L. Lempereur, and S. Maisonneuve, Biologicals44, 139–149 (2016).
- M. Schnodt and H. Buning, Hum. Gene Ther. Methods28, 101–108 (2017).
- E. Burova and E. Ioffe, Gene Ther. 12 Suppl 1, S5–17 (2005).
- E. Lecomte, B. Tournaire, B. Cogne, J.B. Dupont, P. Lindenbaum, M. Martin-Fontaine, F. Broucque, C. Robin, M. Hebben, O.W. Merten, V. Blouin, A. Francois, R. Redon, P. Moullier, and A. Leger, Mol. Ther. Nucleic Acids 4, e260 (2015).
- B.L. Karger and A. Guttman, Electrophoresis30 Suppl 1, S196–202 (2009).
- M. Szarka and A. Guttman, Anal. Chem.89, 10673–10678 (2017).
Jane Luo is a staff applications scientist in the Applied Biologics Workflow Development group at Sciex. She earned her PhD in Biochemistry from the City University of New York, USA, received postdoctoral training on Molecular Biology and Cell Biology at Weill Cornell Medical College and Harvard Medical School, and performed cancer research as an assistant adjunct professor at UC Irvine, USA. In 2002, she moved to industry to develop capillary electrophoresis‑based products and applications.
András Guttman is a senior manager in the separation application team at Sciex, also a professor at the University of Debrecen, Hungary, mostly focusing on capillary electrophoresis analysis of new therapeutic modalities. During his 30 years as a separation scientist he has published more than 360 peer reviewed articles and book chapters, has given more than 400 presentations worldwide and has received numerous awards.
Direct correspondence to: amatheson@mjhlifesciences.com















Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.


Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.

