In Search of The Needle in The Mariana Trench: Host Cell Proteins and the Problem of Dynamic Range
This article explores the analytical challenges associated with HCP monitoring and reviews recent advances in HCP characterization, with special emphasis on high performance liquid chromatography mass spectrometry (HPLC–MS)-based methods and HCP enrichment techniques.
In the course of biopharmaceutical production, host cell proteins (HCPs) may end up in the final drug product despite extensive purification processes. Since these contaminants potentially affect the efficacy and safety of a drug, regulatory guidelines require their levels to be as low as technically feasible. Consequently, the low abundance of HCPs relative to the drug substance (typically, below 100 ppm) poses a key challenge for analytical methods, which must cover a wide dynamic range of up to six orders of magnitude. Here, we outline the root of the analytical problem and review recent advances in HCP characterization, with special emphasis on high performance liquid chromatography mass spectrometry (HPLC–MS)-based methods and HCP-enrichment techniques.
The production of biopharmaceuticals involves recombinant expression of the therapeutic protein by host cells, followed by extensive protein purification in the course of downstream processing. In this context, residual proteins originating from the expression host are referred to as host cell proteins (HCPs). These process‑related impurities may affect the quality and efficacy of a drug product as well as patient safety (1). Consequently, they are regarded in the context of critical quality attributes, that is, “physical, chemical, biological or microbiological propert[ies] or characteristic[s] that should be within an appropriate limit, range, or distribution to ensure the desired product quality” (2). Regulatory guidelines require that HCP levels are as low as technically feasible, but do not explicitly specify acceptable limits (3,4). Hence, manufacturers typically employ a risk-based approach (5) and aim at reducing HCP levels below 100 ng of HCP per mg of drug substance (that is, below 100 ppm) (6). These requirements entail a key challenge in HCP analysis: Due to the desired low abundance of HCPs relative to the drug substance, suitable analytical methods must cover a wide dynamic range of up to six orders of magnitude (Figure 1).


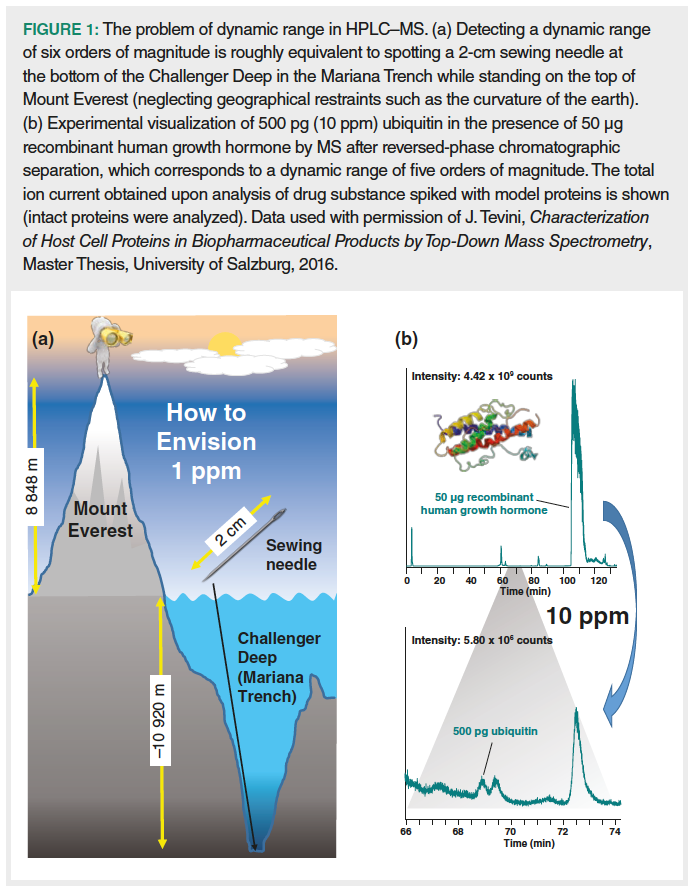
Typically, detection schemes for protein analysis such as UV‑spectroscopy or mass spectrometry (MS) are able to cover a dynamic range of only three to four orders of magnitude. Methodological approaches to enhance the dynamic range of protein detection include (i) highly selective detection, for example, based on immuno‑reaction with selective antibodies, (ii) suppression of compounds interfering with HCP detection, for instance, by selected reaction monitoring mass spectrometry (SRM-MS) or computer‑based, targeted exclusion of mass spectrometric detection, (iii) increasing the mass loading capacity of separation systems and/or better separation of protein components in the sample mixture, such as by multidimensional, chromatographic separation, and (iv) enrichment of HCPs using selective sample preparation methods such as ultrafiltration or protein A chromatography.
Overall, HCP research deals with two main questions: (i) What is the total amount of HCPs in a drug product? (ii) Which HCPs are present in a drug product? (And—if experimentally accessible—what are their quantities?). While initial studies were almost exclusively concerned with measuring total amounts of HCPs, contemporary research aims at obtaining a more detailed view on the HCP profile of a drug product, which culminates in the identification of individual HCPs and their quantification. On the one hand, this shift of interest was facilitated by technological advances—in essence, the emergence of MS-based methods (7). On the other hand, it was recognized that certain HCPs are more difficult to remove than others during purification of the drug substance (8–10) and that, on rare occasions, even a single HCP may compromize the quality of a drug product (11,12) or provoke an immune response upon its administration (13).
In line with these evolving research questions, the repertoire of analytical methods for HCP characterization has greatly increased. Traditionally, the total amount of HCPs in a sample was evaluated by immunospecific methods, such as enzyme-linked immunosorbent assay (ELISA), Western blot, slot blot, and biosensors, while two-dimensional gel electrophoresis was employed for qualitative analyses (see reference [14] for a comprehensive review). However, methods employing MS for HCP identification and quantification have emerged in recent years. Mostly, MS is hyphenated to one- or two‑dimensional high performance liquid chromatography (HPLC); however, setups that adopt capillary electrophoresis for separation have also been described (15–18). In this article, we review recent developments in HCP analysis in reference to established methodology, with a focus on automation of ELISA, two‑dimensional HPLC–MS, HPLC–MS with selective detection, and HCP-enrichment techniques.
ELISA
For almost four decades, ELISA has been used to obtain the total content of HCPs in biopharmaceuticals; thus, this technique has been called the “workhorse method for HCP testing” (19). ELISA employs polyclonal antibodies (pAbs) raised against the supernatant of null cells (that is, cells that do not express the product gene) as primary antibodies for HCP detection (20–22). pAbs should have a broad specificity, which allows them to cover a large portion of the host cell proteome (23–25). Importantly, sensitivity of ELISA crucially depends on the immunogenicity of each individual HCP in the host system (for example, goat, rabbit) where the pAbs were raised. Consequently, this assay is unable to detect non-immunoreactive HCPs (19).
In order to analyze a large number of samples, ELISA can be performed in a high throughput manner. Rey and Wendeler (26) have described a robotic system that fully automates sandwich ELISA on two 96-well microtiter plates. This system allows processing of 24 samples within 7 h. Recently, van Manen-Brush et al. (27) implemented a new HCP immunoassay technology. This microfluidics platform employs cartridges containing 72 sample wells, each of which is linked to three individual glass nanoreactors, facilitating parallel measurements in triplicate. Precision and accuracy of this new technology was found to be comparable to those of traditional ELISA; moreover, the automated system benefits from reduced sample volumes (25 µL vs. 100 µL per well) and assay time (75 min compared to 7 h).
Two-Dimensional (2D)-HPLC–MS
HPLC–MS is a relatively recent addition to the HCP analysis toolbox and orthogonal to immunospecific methods, because it does not rely on specific pAbs for detection. Instead of reporting a total HCP content, individual HCPs may be identified in a bottom-up approach, which generally comprises (i) proteolytic digestion of a sample containing HCPs; (ii) separation of the generated peptides by HPLC; and (iii) peptide identification by tandem MS using an HCP sequence database. However, this simple strategy has several drawbacks: First, its dynamic range is limited to three to four orders of magnitude, which is too narrow for analysis of low abundant HCPs (28). Second, drug substance- and HCP‑derived peptides may co‑elute, which results in ion suppression of low abundant HCP peptides and prevents their detection (29,30).
Two-dimensional HPLC represents one possibility to overcome these limitations. Setups for HCP analysis (31–38) generally employ reversed‑phase (RP) separation in both dimensions, which differ in the pH value of the mobile phase (typically, pH 10 in the first and pH 2.5 in the second dimension). A widely used online 2D‑HPLC system by Doneanu et al. (31) and Farrell et al. (37) combines these RP dimensions with a quadrupole‑time of flight mass spectrometer (QTOF-MS) operated in MSE mode. (MSE is a data‑independent acquisition [DIA] mode, which simultaneously detects peptide precursor and the corresponding fragment ions by alternating between low and high collision energy [39].) This setup lowers the detection limit to 50 ppm and allows label‑free quantification of HCPs via the so-called Hi3 method, where the added MS signals of the three best responding peptides of each protein are compared to the signal of the top three peptides of spiked standard proteins (40). In this way, 33 HCPs could be quantified upon tryptic digestion of samples of PTG1, a chimeric anti‑phosphotyrosine IgG1 monoclonal antibody (mAb).
This prototypic system has been modified in different ways. For example, Doneanu et al. (35) were able to further decrease the limit of quantification down to 1 ppm for HCPs in National Institute of Standards and Technology Monoclonal Antibody Reference Material 8671 (NISTmAb) and Infliximab by the following improvements: (i) Charge surface‑modified C18 columns that minimized peak distortion allowed to load five times more sample (that is, 500 µg). (ii) A travelling‑wave ion mobility (TWIM) cell introduced a “third” dimension that separated co‑eluting peptide precursor ions in the gas phase. Thus, the complexity of MS spectra was further reduced, which in turn enhanced detection of low‑abundant precursors. (iii) The collision energy for fragmentation of precursor ions was correlated with their mobility drift time, which improved fragmentation efficiency for low-abundant HCP peptides. The reproducibility of this system could be established via an interlaboratory study, in which 27 out of 34 NISTmAb HCPs were identified by at least two sites, and 14 by all three labs.
Based on the 2D‑HPLC system introduced by Doneanu et al. (31), Yang et al. (38) employed offline coupling with concatenation of fractions after the first dimension, that is, 60 fractions were pooled to 10 fractions by combining every sixth collected fraction with equal time intervals. This limited the number of samples for the second dimension while retaining separation of peptides with similar retention times in the first dimension. Thereby, they achieved a reliable limit of detection of 10 ppm.
HPLC–MS with Selective Detection
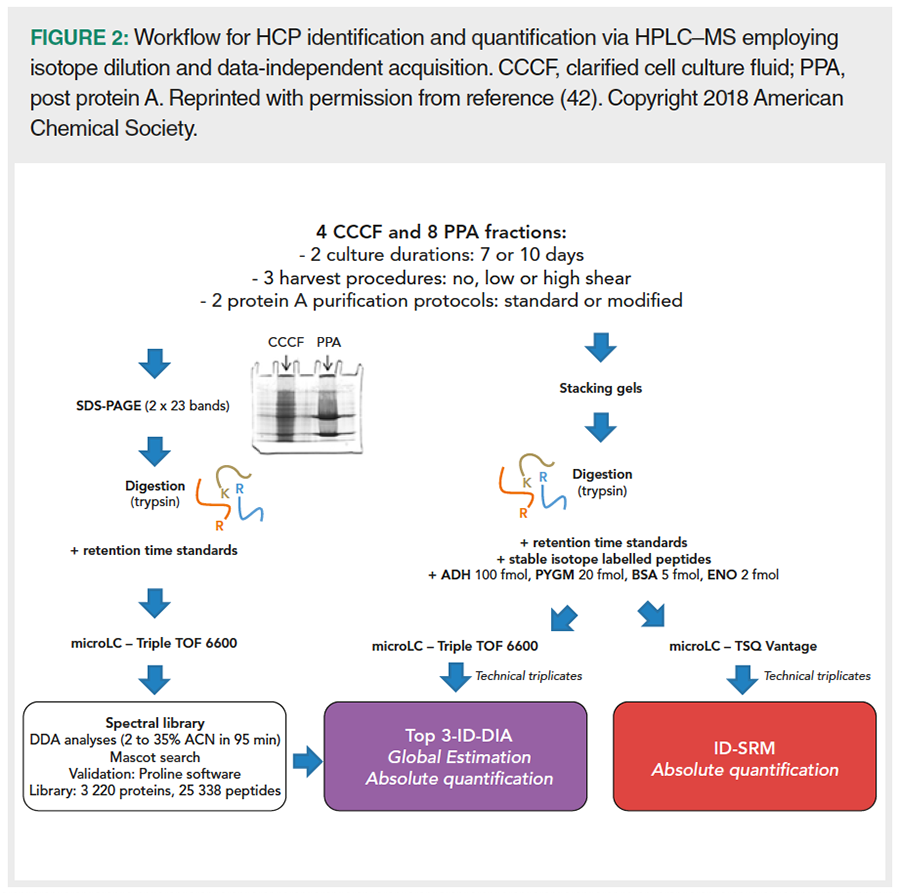
As an alternative to two‑dimensional systems, HPLC–MS setups that use a single chromatographic dimension have been developed, which reduces time but typically requires advanced MS acquisition modes and data evaluation strategies. Kreimer et al. (41) devised a four‑stage HCP identification and quantification assay. In stage 1, an assay library was prepared via 2D‑HPLC–MS and top 15 data‑dependent acquisition (DDA). For each peptide, this library comprised normalized retention time, precursor m/z, and relative intensities of characteristic fragment ions. In stages 2 and 3, one‑dimensional HPLC–MS with DIA and targeted as well as untargeted data evaluation strategies yielded 154 putative HCP and standard peptide identifications in a purified IgG1‑type mAb in the early preclinical stages of development. In the final step, these identifications were verified by parallel reaction monitoring, which allowed Kreimer et al. to identify and quantify all standards and 16 HCPs down to the low ppm level. Husson et al. (42) presented a similar workflow to monitor the impact of culture duration, harvest procedures, and protein A purification protocols on HCP content of an IgG4‑type mAb (Figure 2). Top 3-isotope dilution (ID)‑DIA allowed to identify 1454 HCPs on average in clarified cell culture fluid (with individual amounts between 0.5 and 16192 ppm), and 119 HCPs in post‑protein A fractions (2646 to 5386 ppm). Thus, a dynamic range of five orders of magnitude was achieved. In addition, ten selected peptides were absolutely quantified by ID‑DIA and compared to values from quantification via ID combined with selected reaction monitoring. Results confirmed that these methods were equivalent for HCP quantification, while ID‑DIA was cheaper and required less method setup. If only selected HCPs are required to be tracked over several batches of biopharmaceuticals or during process development, setups employing one‑dimensional HPLC and TWIM with multiple reaction monitoring (MRM) (43) or multiplex MRM (44) are also available.

HCP Enrichment
HCP enrichment (or, equivalently, drug substance depletion) techniques introduce an additional sample preparation step to increase HCP abundances relative to the abundance of the drug substance. The best‑known enrichment strategy makes use of immobilized protein A, a cell wall protein of Staphylococcus aureus that specifically interacts with the Fc domain of mAbs and Fc‑fusion proteins (45,46). Consequently, protein A affinity chromatography (PAAC) not only facilitates purification of Fc domain‑containing biopharmaceuticals from cell culture fluids, but also removal of excess drug substance prior to HCP characterization. Thereby, only a single chromatographic dimension is required during HPLC–MS analysis (47). In addition, separate analyses of PAAC flow‑through and wash fractions reveal HCPs that interact with the drug substance or PAAC resin (48–50). Since protein A‑coupled resins are commercially available and the method is generally simple to use, it has been widely adopted. Alternatives to protein A for selective removal of drug substance include protein L from Peptococcus magnus, which binds kappa light chains in the Fab domain (51), or pAbs that are usually employed in ELISA but can also be coupled to resin material (52). Reisinger et al. (53) further improved HCP identification in protein A‑depleted samples by supplementing both an exclusion and inclusion mass list for MS data recording. The exclusion mass list prevented peptide ions derived from the drug substance from being fragmented, while the inclusion mass list ensured that peptide ions derived from HCPs were preferentially (but not exclusively) fragmented.
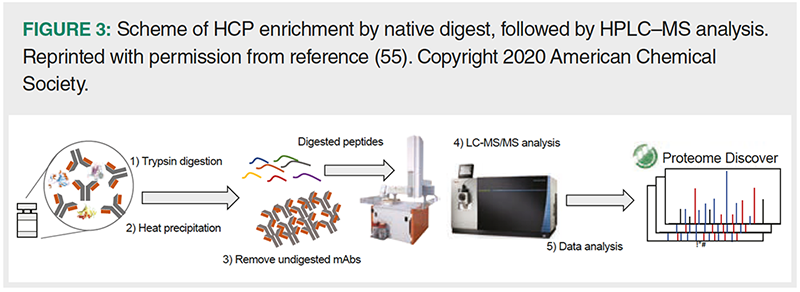
Huang et al. (54) developed a “native digest”, which involves tryptic digestion of HCP- and mAb‑containing samples under native conditions (that is, 25 mM Tris, pH 8) overnight (Figure 3). These mild conditions preserve the native state of the mAb. Hence, it remains largely intact or is only cleaved in the hinge region, as observed for immunoglobulins of subclass IgG4 and IgG1, respectively. While almost no mAb peptides were generated, the majority of HCPs were digested under the applied conditions, except for HCPs containing a large number of cysteine residues. Following digestion, samples were reduced, and intact mAb was precipitated by heating and removed by centrifugation. This sample preparation procedure confined the required dynamic range to 3–4 orders of magnitude. Using native digest, 59 HCPs were detected in NISTmAb by at least two unique peptides in all of the three replicates. Furthermore, the method has been improved with respect to instrument time (21 min compared to previously 8 h) (55) and the number of identified HCPs (602 vs. 55 HCPs) (56).

Chen et al. (57) developed molecular weight cutoff (MWCO) enrichment: First, HCPs were dissociated from the mAb in a buffer containing anionic detergents. Subsequently, samples were filtered over a 50 kDa MWCO membrane. Since 80% of HCPs derived from Chinese hamster ovary cells are smaller than 75 kDa (compared to the mAb of approximately 150 kDa), this procedure efficiently separated HCPs from the mAb and allowed the identification of 164 HCPs in NISTmAb via standard tryptic digestion followed by HPLC–MS analysis.
Several other HCP enrichment strategies have been described. Wang et al. (58) applied hydrophilic interaction chromatography, where a wide pore (300 Å) amide column packed with 1.7 µm particles facilitated injection of 700 µg sample. Three fractions were collected from a water/acetonitrile gradient. The second fraction comprised a large peak corresponding to the drug substance, whereas the bulk of HCPs eluted in the first fraction. In this way, 71 HCPs were identified in NISTmAb. Mortstedt et al. enriched HCPs using instrumentation that employs a bead‑based library of combinatorial hexapeptide ligands (59). If a sample containing the drug substance and HCPs is applied to these beads, binding sites for the high abundant drug substance are rapidly saturated, whereas low abundant HCPs are concentrated. Some HCP identification workflows include a peptide fractionation step between tryptic digestion and HPLC–MS analysis (55,60).
Conclusion
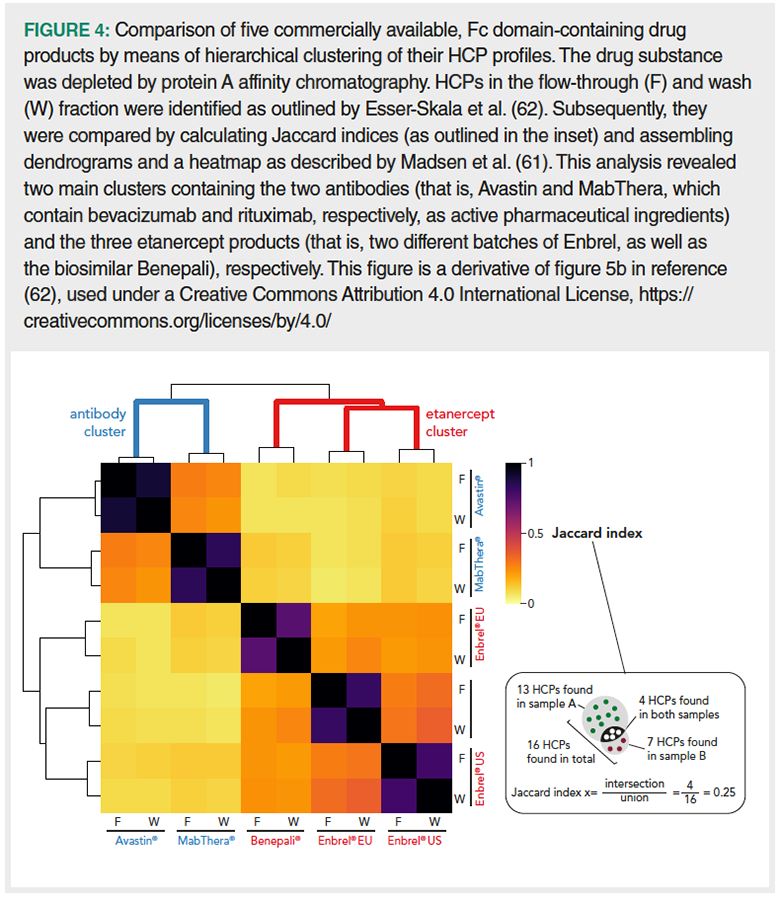
Both academic and industrial scientists have an ever‑growing repertoire of methods for HCP characterization at their disposal. Sophisticated 2D-HPLC‑MS-based setups, 1D-HPLC‑MS with selective detection, as well as simple HCP enrichment techniques cover a broad range of HCP research topics with varying degrees of granularity. Application of current methodologies ensures that total HCP levels in a drug product are sufficiently low; moreover, it facilitates monitoring of individual HCPs that are known to be particularly problematic, thereby increasing drug safety. Furthermore, comprehensive knowledge on the HCP content of a sample promotes advanced data analysis strategies (and vice versa), such as hierarchical clustering for in‑depth comparison of HCP profiles of different cell lines, upstream and downstream processes (61) or commercially available drug products (62) (Figure 4). Certainly, technological advances will provide increased sensitivity in the future, which will boost visibility of the “needle in the Mariana Trench”.

Acknowledgements
The financial support by the Austrian Federal Ministry of Digital and Economic Affairs, the National Foundation of Research, Technology and Development, as well as a Start‑up Grant of the State of Salzburg is gratefully acknowledged.
Conflicts of Interest
Novartis BTDM, Sandoz GmbH as well as Thermo Fisher Scientific provide financial support for the Christian Doppler Laboratory for Innovative Tools for Biosimilar Characterization. The salaries of Wolfgang Esser-Skala and Therese Wohlschlager are fully funded; Christian G. Huber’s salary is partly funded by the Christian Doppler Laboratory for Biosimilar Characterization. The authors declare no other competing financial interest.
References
- M. Vanderlaan, J. Zhu-Shimoni, S. Lin, F. Gunawan, T. Waerner, and K.E. Van Cott, Biotechnol. Prog. 34, 828–837 (2018).
- European Medicines Agency. ICH guideline Q8 (R2) on pharmaceutical development, 2017.
- European Medicines Agency. ICH Topic Q 6 B. Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products, 2006.
- U.S. Pharmacopeial Convention. (1132) Residual Host Cell Protein Measurement in Biopharmaceuticals, 2016.
- D.G. Bracewell, R. Francis, and C.M. Smales, Biotechnol. Bioeng. 112, 1727–1737 (2015).
- C.L. de Zafra, V. Quarmby, K. Francissen, M. Vanderlaan, and J. Zhu-Shimoni, Biotechnol. Bioeng. 112, 2284–2291 (2015).
- R. Aebersold and D.R. Goodlett, Chem. Rev. 101, 269–295 (2001).
- Q. Zhang, A.M. Goetze, H. Cui, J. Wylie, B. Tillotson, A. Hewig, M.P. Hall, and G.C. Flynn, Biotechnol. Prog. 32, 708–717 (2016).
- K.N. Valente, N.E. Levy, K.H. Lee, and A.M. Lenhoff, Curr. Opin. Biotechnol. 53, 144–150 (2018).
- S.K. Singh, A. Mishra, D. Yadav, N. Budholiya, and A.S. Rathore, Biotechnol. Prog. 36, e2936 (2020).
- S.X. Gao, Y. Zhang, K. Stansberry-Perkins, A. Buko, S. Bai, V. Nguyen, and M.L. Brader, Biotechnol. Bioeng. 108, 977–982 (2011).
- T. Hall, S.L. Sandefur, C.C. Frye, T.L. Tuley, and L. Huang, J. Pharm. Sci. 105, 1633–1642 (2016).
- S.K. Fischer, M. Cheu, K. Peng, J. Lowe, J. Araujo, E. Murray, D. McClintock, J. Matthews, P. Siguenza, and A. Song, AAPS J. 19, 254–263 (2017).
- A.L. Tscheliessnig, J. Konrath, R. Bates, and A. Jungbauer, Biotechnol. J. 8, 655–670 (2013).
- G. Zhu, L. Sun, R. Wojcik, D. Kernaghan, J.B. McGivney IV, and N.J. Dovichi, Talanta 98, 253–256 (2012).
- G. Zhu, L. Sun, T. Linkous, D. Kernaghan, J.B. McGivney IV, and N.J. Dovichi, Electrophoresis 35, 1448–1452 (2014).
- G. Zhu, L. Sun, J. Heidbrink-Thompson, S. Kuntumalla, H.Y. Lin, C.J. Larkin, J.B. McGivney IV, and N.J. Dovichi, Electrophoresis 37, 616–622 (2016).
- Z. Zhang, T. Albanetti, T. Linkous, C.J. Larkin, R. Schoner, J.B. McGivney IV, and N.J. Dovichi, Electrophoresis 38(3–4), 401–407 (2017).
- J. Zhu-Shimoni, C. Yu, J. Nishihara, R.M. Wong, F. Gunawan, M. Lin, D. Krawitz, P. Liu, W. Sandoval, and M. Vanderlaan, Biotechnol. Bioeng. 111, 2367–2379 (2014).
- V.R. Anicetti, E.F. Fehskens, B.R. Reed, A.B. Chen, P. Moore, M.D. Geier, and A.J. Jones, J. Immunol. Methods 91(2), 213–224 (1986).
- L.C. Eaton, J. Chromatogr. A. 705(1), 105–114 (1995).
- N. Dagouassat, J.F. Haeuw, V. Robillard, F. Damien, C. Libon, N. Corvaia, F. Lawny, T.N. Nguyen, J.Y. Bonnefoy, and A. Beck, J. Immunol. Methods 251, 151–159 (2001).
- C.S. Lundstrom, A. Mattsson, K. Lovgren, A. Eriksson, A.L. Sundberg, M. Lundgren, and C.E. Nilsson, Vaccine 32(24), 2911–2915 (2014).
- P.A. Baldus, M. Brown, R.S. Wright, J.A. Campbell, S. Lute, B. Chavez, K. Brorson, and N. Mozier, Biotechnol. Bioeng. 115(2), 413–422 (2018).
- K. Pilely, S.B. Nielsen, A. Draborg, M.L. Henriksen, S.W.K. Hansen, L. Skriver, E. Mørtz, and R.R. Lund, Biotechnol. Prog. 36(4), e2983 (2020).
- G. Rey and M.W. Wendeler, J. Pharm. Biomed. Anal. 70, 580–586 (2012).
- K. van Manen-Brush, J. Zeitler, J.R. White, P. Younge, S. Willis, and M. Jones, Biotechniques 69 (2020).
- L. Wu and D.K. Han, Expert Rev. Proteomics 3, 611–619 (2006).
- B.K. Matuszewski, M.L. Constanzer, and C.M. Chavez-Eng, Anal. Chem. 75(13), 3019–3030 (2003).
- A. Michalski, J. Cox, and M. Mann, J. Proteome Res. 10(4), 1785–1793 (2011).
- C.E. Doneanu, A. Xenopoulos, K. Fadgen, J. Murphy, S.J. Skilton, H. Prentice, M. Stapels, and W. Chen, MAbs 4(1), 24–44 (2012).
- M.R. Schenauer, G.C. Flynn, and A.M. Goetze, Anal. Biochem. 428(2), 150–157 (2012).
- M.R. Schenauer, G.C. Flynn, and A.M. Goetze, Biotechnol. Prog. 29, 951–957 (2013).
- Q. Zhang, A.M. Goetze, H. Cui, J. Wylie, S. Trimble, A. Hewig, and G.C. Flynn, MAbs 6(3), 659–670 (2014).
- C.E. Doneanu, M. Anderson, B.J. Williams, M.A. Lauber, A. Chakraborty, and W. Chen, Anal. Chem. 87(20), 10283–10291 (2015).
- K. Sandra, A. Ortiz, and P. Sandra, LCGC Europe Supplement: Advances in Biopharmaceutical Analysis 28(s10), 45–48 (2015).
- A. Farrell, S. Mittermayr, B. Morrissey, N. Mc Loughlin, N. Navas Iglesias, I.W. Marison, and J. Bones, Anal. Chem. 87(18), 9186–9193 (2015).
- F. Yang, D.E. Walker, J. Schoenfelder, J. Carver, A. Zhang, D. Li, R. Harris, J.T. Stults, X.C. Yu, and D.A. Michels, Anal. Chem. 90(22), 13365–13372 (2018).
- R.H. Bateman, R. Carruthers, J.B. Hoyes, C. Jones, J.I. Langridge, A. Millar, and J.P. Vissers, J. Am. Soc. Mass Spectrom. 13(7), 792–803 (2002).
- J.C. Silva, M.V. Gorenstein, G.Z. Li, J.P. Vissers, and S.J. Geromanos, Mol. Cell. Proteomics 5(1), 144–156 (2006).
- S. Kreimer, Y. Gao, S. Ray, M. Jin, Z. Tan, N.A. Mussa, L. Tao, Z. Li, A.R. Ivanov, and B.L. Karger, Anal. Chem. 89(10), 5294–5302 (2017).
- G. Husson, A. Delangle, J. O’Hara, S. Cianferani, A. Gervais, A. Van Dorsselaer, D. Bracewell, and C. Carapito, Anal. Chem. 90(2), 1241–1247 (2018).
- C. Doneanu, J. Fang, Y. Alelyunas, Y.Q. Yu, M. Wrona, and W. Chen, J. Vis. Exp. 134, e55325 (2018).
- X. Gao, B. Rawal, Y. Wang, X. Li, D. Wylie, Y.-H. Liu, L. Breunig, D. Driscoll, F. Wang, and D.D. Richardson, Anal. Chem. 92(1), 1007–1015 (2020).
- H. Hjelm, K. Hjelm, and J. Sjöquist, FEBS Lett. 28(1), 73–76 (1972).
- S. Hober, K. Nord, and M. Linhult, J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 848(1), 40–47 (2007).
- J.H. Thompson, W.K. Chung, M. Zhu, L. Tie, Y. Lu, N. Aboulaich, R. Strouse, and W.D. Mo, Rapid Commun. Mass Spectrom. 28(8), 855–860 (2014).
- N. Aboulaich, W.K. Chung, J.H. Thompson, C. Larkin, D. Robbins, and M. Zhu, Biotechnol. Prog. 30(5), 1114–1124 (2014).
- S. Chollangi, R. Parker, N. Singh, Y. Li, M. Borys, and Z. Li, Biotechnol. Bioeng. 112(11), 2292–2304 (2015).
- A.A. Shukla and P. Hinckley, Biotechnol. Prog. 24(5), 1115–1121 (2008).
- L. Bjorck, J. Immunol. 140(4), 1194–1197 (1988).
- K. Bomans, A. Lang, V. Roedl, L. Adolf, K. Kyriosoglou, K. Diepold, G. Eberl, M. Molhoj, U. Strauss, C. Schmalz, R. Vogel, D. Reusch, H. Wegele, M. Wiedmann, and P. Bulau, PLoS One 8, e81639 (2013).
- V. Reisinger, H. Toll, R.E. Mayer, J. Visser, and F. Wolschin, Anal. Biochem. 463, 1–6 (2014).
- L. Huang, N. Wang, C.E. Mitchell, T. Brownlee, S.R. Maple, and M.R. De Felippis, Anal. Chem. 89(10), 5436–5444 (2017).
- J. Ma and G.W. Kilby, J. Proteome. Res. 19(8), 3396–3404 (2020).
- R.O. Johnson, T. Greer, M. Cejkov, X. Zheng, and N. Li, Anal. Chem. 92(15), 10478–10484 (2020).
- I.-H. Chen, H. Xiao, T. Daly, and N. Li, Anal. Chem. 92(5), 3751–3757 (2020).
- Q. Wang, T.R. Slaney, W. Wu, R. Ludwig, L. Tao, and A. Leone, Anal. Chem. 92(15), 10327–10335 (2020).
- H. Mörtstedt, Å. Makower, P.-O. Edlund, K. Sjöberg, and A. Tjernberg, J. Pharm. Biomed. Anal. 185, 113256 (2020).
- R. Kufer, M. Haindl, H. Wegele, and S. Wohlrab, Anal. Chem. 91(15), 9716–9723 (2019).
- J.A. Madsen, V. Farutin, T. Carbeau, S. Wudyka, Y. Yin, S. Smith, J. Anderson, and I. Capila, MAbs 7(6), 1128–1137 (2015).
- W. Esser-Skala, M. Segl, T. Wohlschlager, V. Reisinger, J. Holzmann, and C.G. Huber, Anal. Bioanal. Chem. 412, 6583–6593 (2020).
Wolfgang Esser-Skala is a postdoctoral researcher at the Christian Doppler Laboratory for Innovative Tools for Biosimilar Characterization at the University of Salzburg, Austria. He primarily develops algorithms and workflows for analyzing mass spectrometric data on post-translational modifications found in biopharmaceuticals.
Therese Wohlschlager is a postdoctoral researcher at the University of Salzburg, Austria. Within the Christian Doppler Laboratory for Biosimilar Characterization, she focuses on the development of analytical tools for the characterization of glycosylated biopharmaceuticals.
Christian G. Huber trained as an analytical chemist at the University of Innsbruck, Austria, and is currently a professor of chemistry for biosciences at the University of Salzburg. He also leads the Christian Doppler Laboratory for Biosimilar Characterization, which cooperates with Novartis and Thermo Fisher Scientific. His research interests include proteome and metabolome analysis as well as in-depth protein characterization by means of HPLC and MS.













Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.


Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

