Exploring Capillary Electrophoresis–Mass Spectrometry for the In-Depth Intact Native Analysis of Cysteine-Conjugated Antibody-Drug Conjugates
This article explores the potential of microchip capillary electrophoresis–mass spectrometry (CE–MS) for the characterization of intact biopharmaceuticals, and how this technique can effectively be employed to monitor essential quality attributes of interest.
Monoclonal antibodies are produced in mammalian cell culture and thus possess a considerable complexity due to variable N-glycosylation and other, up- and downstream processing induced modifications (1). Related therapeutic formats like Fc-fusion proteins and antibody‑drug conjugates (ADCs) show additional layers of complexity, either derived from higher levels of N- and O‑glycosylation or in the case of ADCs, the presence of varying numbers of small cytotoxic compounds attached to the protein scaffold, which acts as a drug delivery system. In‑depth analysis of the primary protein sequence as well as amino acid modifications is routinely performed by peptide mapping. However, peptide mapping fails to reveal the molecular structure and assembly on the intact protein level based on the use of proteases that digest the protein into peptides prior to analysis. Recent technological advances in separation science and high-resolution mass spectrometry (MS) have greatly expanded the utility of intact protein MS. Direct MS interfacing of highly selective separations based on molecule size (2), charge (3–6) and hydrophobicity (7–9) has been shown to be possible. These separation modes operate under physiological conditions and thus preserve the higher order structure of proteins (10). Thereby, when combined with native high-resolution mass spectrometry, they allow for the analysis of molecules of increasing complexity as a result of the chromatographic or electrophoretic separation of protein isoforms in combination with greater spatial mass spectral resolution in the higher m/z range that is characteristic of native MS. Moreover, the native analysis conditions facilitate characterization of highly labile molecules such as cysteine-conjugated ADCs (11).
The characterization of ADCs at the intact protein level is important for the determination of the average drug‑antibody ratio (DAR). This measure determines how much cytotoxic compound is administered on average per protein molecule and hence, is directly related to the clinical outcome.
Here, we employed microfluidic chip‑based capillary electrophoresis–mass spectrometry (CE–MS) for the characterization of a cysteine conjugated ADC mimic (6). Characterization was performed before and after enzymatic deglycosylation using two different acquisition modes. Firstly, via the infusion option which allows for static nanospray infusion of analytes when only pressure is applied across the microchip channel, and secondly, via charge variant separation prior to MS detection when the separation voltage is applied. The MS data obtained was then analysed to investigate whether the native‑like protein structure and non‑covalent interactions between conjugated heavy and light chains were maintained. Both approaches were compared to evaluate the depth of information gathered including information on the average DAR and other features such as N-glycosylation and charge sensitive modifications.
Materials and Methods
Chemicals and Consumables: The ADC mimic (MSQC8) was obtained from Sigma-Aldrich and consists of the SILu Lite SigmaMAb (MSQC4) conjugated to dansyl fluorophores via a LC-SMCC (succinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxy-[6-amidocaproate]) crosslinker. MS grade water (Optima) was purchased from Fisher. PNGase F (Carbo Clip) was obtained from Asparia Glycomics. Vivaspin 500 10 kDa spin filters were obtained from Sartorius. The ZipChip Native Antibodies Kit and HRN chips were provided by 908 Devices. LC–MS grade dimethyl sulfoxide (DMSO) was obtained from Thermo Fisher Scientific.
Sample Preparation: For deglycosylation, 2.5 µL of PNGase F were added to 100 µg of glycoprotein and the reaction was maintained at 37 °C for 180 min under constant agitation at 400 rpm. Both glycosylated and deglycosylated samples were buffer exchanged using 10 kDa Vivaspin spin filters. Sample diluent was intact native background electrolyte, consisting of an ammonium acetate based buffer at pH 5.5 containing 4% DMSO (v/v) (12).
A final sample concentration of 0.1 mg/mL was used for infusion experiments and 1.0 mg/mL for charge variant separations.
Instrumentation, Sample and Data Analysis: For CE–MS experiments, the 908 Devices ZipChip interface, equipped with a HRN chip and connected to a model 840 autosampler was attached to a Q Exactive Plus hybrid quadrupole‑Orbitrap instrument equipped with the extended mass biopharma option (Thermo Fisher Scientific). To increase the sensitivity, the ZipChip background electrolyte contained 4% DMSO (v/v), while a constant field strength of 500 V/cm was applied for separation. Charge variant separations were based on the injection of 1 nL sample. MS data acquisition was performed in High Mass Range (HMR) mode. All analyses were performed at a resolution setting of 35 000 (at m/z 200) and an in‑source collision induced dissociation (CID) value of 100 eV, the number of microscans was 5 and AGC target and maximum inject time were 5e5 and 50, respectively. Direct infusion data was acquired using the averaging function. Spectral deconvolution was performed using the ReSpect algorithm (Thermo Fisher Scientific). Deconvolution and further data analysis were performed in BioPharma Finder software version 3.2 (Thermo Fisher Scientific).
Nano Infusion of ADC Mimic Before and After Deglycosylation
A requirement for the intact analysis of cysteine conjugated ADCs is to maintain conditions during analysis that prevent the dissociation of the conjugated heavy and light antibody chains. The microchip CE–MS system operated using a background electrolyte that is designed to maintain proteins and protein complexes in their native state. Besides the capabilities for CE–MS, the microchip device can also be used for nanospray infusion by applying pressure on the microchannel. Figure 1(a) shows the charge envelope obtained for direct infusion of the ADC mimic. Evidently, the major charge state z=+24 is situated close to m/z 6,000. For a molecule with a mass of 150 kDa, this clearly infers preservation of the higher order structure (13). A zoom into charge state z=+24 shown on the right-hand side of Figure 1(a) shows the heterogeneity derived from conjugation with up to eight payloads as well as the glycan heterogeneity within the DAR‑distribution. Both the appearance in the high m/z range as well as the DAR distribution demonstrates that labile inter‑chain interactions are being maintained during the analysis. Figure 1(b) shows the appearance of the MS raw data after deglycosylation of the ADC mimic. The charge envelope is expanded and lower charge states become more prevalent, which is likely due to an exposure of additional charged residues by the deglycosylation process. The right panel of Figure 1(b) shows a similar distribution in terms of DAR but with a shift towards lower m/z and without the heterogeneity caused by various N-glycosylation.



Deconvolution of the spectra from Figure 1(a) yields a distribution of peaks in five distinct clusters (Figure 2). Clusters represent DAR forms which differ by 1,336 Da, representing the mass shift caused by two payloads of 668 Da each. Hence, the five clusters represent DAR 0, 2, 4, 6, and 8 forms with varying abundance. The calculated average DAR is 4.2, which agrees well with the manufacturer’s specifications (14). Additionally, the glycoforms and their relative abundances can be obtained for each DAR form. The most abundant peak represents the glycoform pair G0F/G1F followed by G0F/G0F and G1F/G1F or G0F/G2F. In general, experimental masses are in excellent agreement with the theoretical masses calculated. For example, mass deviations of the mentioned glycoforms of the DAR 4 variant are all below 10 ppm, thereby generating high confidence in the data and its associated assignment.


Deglycosylation greatly reduced the overall spectral complexity, as shown in the deconvoluted spectrum in Figure 3. The DAR distribution is similar to what was observed in Figure 2, however the calculated average DAR of 4.0 is slightly lower. Reducing glycan heterogeneity can simplify DAR determination and data analysis, but also has the potential to reveal formerly hidden modifications. As depicted in Figure 3, the main peak is accompanied by two peaks, one in the lower and the other one in the higher mass range. These species are deviating by -318 and +162 Da and represent isoforms derived from non-glycosylation‑related modifications. Peptide mapping experiments revealed the occurrence of glycation events on several lysine residues which is likely the cause for the mass shift of +162 Da observed on the intact level. The -318 Da mass shift was seen in both, glycosylated and deglycosylated data sets. Also in this case, peptide mapping was allocating this modification to a specific site. The conjugated peptide of the antibody light chain was found with two modifications, either +668 Da representing the intact payload or +350 Da, representing the payload with a negative mass offset of 318 Da. Hence, this modification very likely represents a modified form of the payload, most probably deriving from hydrolysis of the amide bond leading to the release of the dansylcadaverine.


Charge Variant Separation of ADC Mimic Before and After Deglycosylation
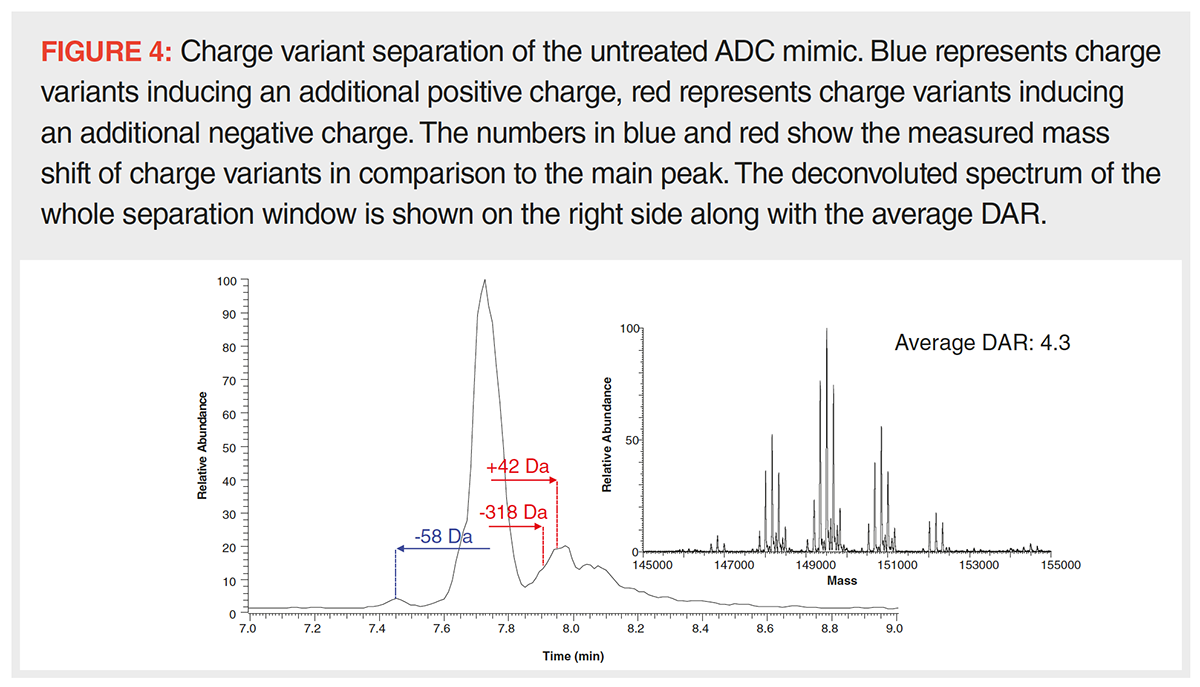
The CE–MS platform used herein allows for the separation of intact protein charge variants with direct MS detection and based on operation at flow rates in the nL/min range and a sample consumption of only around 1 ng per run offers excellent sensitivity. Like strong cation exchange chromatography, CE separates molecules based on differences in their net surface charge, thus, resulting in similar charge variant patterns, albeit with a reversed elution order. Nonetheless, the fundamentally different separation mechanism, which is based on varying migration velocities within an electric field, can result in a different separation selectivity and in different information content (15). The charge variant profile shown in Figure 4 clearly indicates the separation of the main species and several lower abundant charge variants. Such can be seen in front of the main peak, which are caused by modifications inducing a positive or depleting a negative charge, and after the main peak with modifications having the opposite effect. Several charge variants can be found including species with mass offsets of -58, +42, and -318 Da. The variant bearing a payload modification with a mass shift of -318 Da was found to migrate significantly slower than the main species, which clearly suggests a lower positive net charge. The other charge variants were not previously seen during direct infusion experiments, hence the analysis of such species clearly benefits from charge variant separation. A mass shift of -58 Da and an additional positive charge is strongly indicative of C‑terminal glycine loss and proline amidation on one of the heavy chains, the presence of such species was confirmed by peptide mapping (16). A mass of +42 and an additional negative charge infers acetylation of one of the N-termini which has been shown to be a charge sensitive modification on other proteins (17). Also in this case, peptide mapping strongly supports this assumption as the modification was allocated to the N-terminus of the heavy chain. Importantly, even though charge variant separation is performed, an average DAR well within the specifications is obtained, as is shown in Figure 4 on the right hand side, making this approach fit for the simultaneous determination of the average DAR, the glycan distribution and the charge variant heterogeneity. Obviously, the visibility of the lower abundant DAR forms is dependent on the overall relative abundance of the charge variants under investigation, hence, DAR 0 and 8 may not be detected for low abundant species.

Deglycosylation is drastically reducing sample heterogeneity, which further improves analytical sensitivity. Hence, while information about the glycan distribution is lost, other low abundant modifications may be rendered accessible for analysis. We found that this was also the case for the CE–MS analysis of untreated and deglycosylated ADC mimic. Figure 5 shows the electropherogram of the deglycosylated sample which resembles the trace of the untreated sample shown in Figure 4. The signal intensity of the formerly identified modified species was strongly increased and one additional charge variant was detected showing a positive mass offset of 128 Da. A faster migration than the main species is indicating an additional positive charge. The mass shift of +128 Da paired with the migration behaviour indicates the presence of incompletely cleaved C‑terminal lysine variants which can be found at a relative abundance of <1%. Notably, this modification was only found for the DAR 2 form which is certainly related to the unusual DAR distribution seen in this particular instance. A calculation of the average DAR after sliding window deconvolution yielded a value of only 2.0, much lower than reported in previous experiments and well outside of the specifications (Figure 5, right panel). Further analysis revealed that not only charge variants were separated during the CE–MS run, but also different DAR forms with DAR 0 migrating fastest, followed by DAR 2 and lastly DAR 4. Unlike in previous experiments, DAR 4 was found to be of lower abundance than DAR 2, while DAR 6 and eight forms were not detected at all. Interestingly, this phenomenon was not seen when using the same sample for direct infusion and it only occurred after deglycosylation. A change in the physicochemical properties caused by deglycosylation, such as reduced solubility, likely changed the migration behaviour of especially higher DAR forms, effectively preventing their migration in the CE channel.


Summary and Conclusions
Native separation modes coupled on‑line to MS offer great potential for the characterization of large, heterogeneous, and labile molecules such as cysteine-conjugated antibody‑drug conjugates. Here, we explored the potential of microfluidic CE–MS for the characterization of an ADC mimic both by infusion and via charge variant separation. Direct infusion of both, untreated and deglycosylated samples resulted in clear native protein spectra with a characteristic DAR distribution showing either 0, 2, 4, 6, or 8 payloads. The calculated average DAR was 4.2 and 4.0 for untreated and deglycosylated samples respectively, which is well within the specifications provided by the manufacturer. In case of the untreated sample, the glycan distribution was determined with the most abundant glycan pairs being G0F/G1F, followed by G0F/G0F, G1F/G1F, and G1F/G2F. Mass accuracies were within 10 ppm. The data quality was found to be excellent, which can be attributed to the infusion setup and the possibility to average hundreds of spectra acquired over the course of several minutes. Infusion is operated at nL/min flow scale, and only requires a minimum of 10 µL of sample at a concentration of 0.1 mg/mL and thus can be performed with minimal sample consumption. The CE–MS device has also proven powerful for the separation of multiple charge variants of the ADC mimic. Among those were species with subtle to moderate mass shifts which were not detected in infusion experiments. Hence, charge variant separation offers an additional dimension of information, rendering the average DAR, the glycan distribution and the charge variant heterogeneity accessible to analysis. With the here applied technique, this vast information was acquired within only 15 min of analysis time and with a sample consumption of only 1 ng per injection. In the case of the analysis of the untreated sample, the average DAR was highly similar to that obtained in infusion experiments. Deglycosylation and charge variant separation resulted in increased analysis sensitivity but came at the cost of the distortion of the average DAR. Hence, sample treatment should be performed with care and under consideration of the analysis procedure desired. The CE–MS workflow herein employed showed excellent utility for the analysis of a cysteine‑conjugated ADC mimic and thus offers great potential for the analysis of therapeutic ADCs of similar complexity.
Acknowledgements
The authors gratefully acknowledge Shreya Ahuja and Kevin Ray from MilliporeSigma for providing samples and for their general support. We would also like to acknowledge Thermo Fisher Scientific for instrument access and support.
References
- A.Beck, E. Wagner-Rousset, D. Ayoub, A. Van Dorsselaer, and S. Sanglier-Cianférani, Anal. Chem. 85(2), 715–736 (2013).
- A.C. Lazar, L. Wang, W.A. Blättler, G. Amphlett, J.M. Lambert, and W. Zhang, Rapid Commun. Mass Spectrom. 19(13), 1806–1814 (2005).
- F. Füssl, K. Cook, K. Scheffler, A. Farrell, S. Mittermayr, and J. Bones, J. Anal.Chem. 90(7), 4669–4676 (2018).
- A.O. Bailey, G. Han, W. Phung, P. Gazis, J. Sutton, J.L. Josephs, and W. Sandoval, MAbs 10 (8), 1214–1225 (2018).
- F.F. Ma, F. Raoufi, M.A. Bailly, L. Fayadat‑Dilman, and D. Tomazela, MAbs 12(12), 1763762 (2020).
- E.A. Redman, N.G. Batz, J.S. Mellors, and J.M. Ramsey, Anal. Chem. 87(4), 2264–2272 (2015).
- B. Chen, Z. Lin, A.J. Alpert, C. Fu, Q. Zhang, W.A. Pritts, and Y. Ge, Anal. Chem. 90(12), 7135–7138 (2018).
- Y. Yan, T. Xing, S. Wang, T.J. Daly, and N. Li, J. Pharma. Biomed. Anal. 186, 113313 (2020).
- B. Wei, G. Han, J. Tang, W. Sandoval, and Y.T. Zhang, Anal. Chem. 91(24), 15360–15364 (2019).
- R.H. van den Heuvel and A.J. Heck, Curr. Opin. Chem. Biol. 8(5), 519–526 (2004).
- A. Wakankar, Y. Chen, Y. Gokarn, and F.S. Jacobson, MAbs 3, 161–172 (2011).
- 908devices. In Boosting Sensitivity for Intact Antibody Charge Variant Analysis, 2019.
- R.J. Rose, E. Damoc, E. Denisov, A. Makarov, and A.J.R. Heck, Nature Methods 9(11), 1084–1086 (2012).
- Sigma-Aldrich. In SigmaMAb Antibody Drug Conjugate (ADC) Mimic; Sigma-Aldrich: https://www.sigmaaldrich.com/ (2016).
- F. Füssl, A. Trappe, S. Carillo, C. Jakes, and J. Bones, Anal. Chem. 92(7), 5431–5438 (2020).
- T. Kaschak, D. Boyd, F. Lu, G. Derfus, B. Kluck, B. Nogal, C. Emery, C. Summers, K. Zheng, R. Bayer, A. Amanullah, and B.X. Yan, MAbs 3(6), 577–583 (2011).
- F. Füssl, A. Criscuolo, K. Cook, K. Scheffler, and J. Bones, J. Proteome Res. 18(10), 3689–3702 (2019).
Florian Füssl received his Dr. rer. nat. degree from the University of Salzburg, Austria. Since 2016 he has been a postdoctoral researcher in the Characterization and Comparability Laboratory of Jonathan Bones at NIBRT in Dublin, Ireland. His main focus is the development of novel methods for the characterization of complex biopharmaceuticals using liquid chromatography–mass spectrometry (LC–MS) techniques with special emphasis on intact native protein analysis.
Lisa Strasser holds a PhD from the University of Salzburg, Austria. In 2019 she joined the National Institute of Bioprocessing Research and Training (NIBRT), based in Dublin, Ireland, where she was using LC–MS based proteomics to study host cell proteins. Now she is working on the characterization of adeno‑associated viruses (AAVs) for the purpose of gene therapy.
Sara Carillo is the applications development team leader in the Characterization and Comparability Laboratory in NIBRT, Ireland. Sara completed her PhD in Chemical Science at the University of Naples in 2013 working on bacterial glyco‑conjugates structural characterization. In 2015, Sara joined Jonathan Bones’ research group in NIBRT, working on CHO cell glycome and biotherapeutics characterization.
Jonathan Bones is the Principal Investigator of the NIBRT Characterization and Comparability Laboratory and an Associate Professor in the School of Chemical and Bioprocess Engineering at University College Dublin. He holds a PhD in analytical chemistry from Dublin City University and undertook postdoctoral research with Pauline M. Rudd at NIBRT and Barry L. Karger at the Barnett Institute of Chemical and Biological Analysis at Northeastern University, Boston prior to taking up his current position. His research interests are in the application of separation methods coupled to high resolution mass spectrometry for characterization of biological processes and complex biopharmaceuticals.
Direct correspondence to: amatheson@mjhlifesciences.com













Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.


Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

