Analytical Separation Methods for Therapeutic Oligonucleotides
The diverse nature of oligonucleotide therapeutics leads to the requirement for multiple separation methods to characterize quality attributes. Highly chemically-modified to improve efficacy and resilience to nucleases, they are challenging to analyse using a single method. This article discusses multiple chromatographic separation methods, and the benefits of mass spectrometry (MS).
The therapeutic oligonucleotide industry continues to grow due to increasing regulatory approvals and improvements in medicinal chemistry. The term oligonucleotide therapeutic encompasses a diverse range of oligonucleotide molecular structure and function. Among the types of oligonucleotide therapeutic are antisense and splice-switching oligonucleotides, short interfering RNA (siRNA), aptamers, messenger RNA (mRNA), antagomirs, and immunomodulatory oligonucleotides. Oligonucleotides function to manipulate the expression of proteins associated with disease states or pathogenicity. Currently there are 10 approved pharmaceuticals on the market. The diverse nature of oligonucleotide therapeutics leads to the requirement for multiple separation methods to characterize quality attributes. Chromatographic separation is required for post manufacturing purification, characterization of impurities and structural variants, pharmacokinetic studies, and analysis of drug formulations. As oligonucleotide therapeutics are highly chemically-modified to improve efficacy and resilience to nucleases, they are challenging to analyse using a single method. Multiple chromatographic separations can overcome challenges of broad peak shapes and low resolution between closely related analytes. In addition, mass spectrometry (MS) detection enables higher accuracy when characterizing complex samples.
The use of therapeutic oligonucleotides has gained momentum recently within the pharmaceutical industry as a result of increasing regulatory approvals and improvements in medicinal chemistry. The term “therapeutic oligonucleotide” encompasses many types of nucleic acid structure and function. These oligonucleotides (OGNs) manipulate the expression or function of “proteins of interest”, that is, those associated with disease states or pathogenicity. The vast number of active or completed clinical trials for this drug class suggests that the therapeutic OGN industry will continue to grow. Additionally, in the event of a global pandemic, the development of ribonucleic acid (RNA)‑based vaccines for SARS‑CoV‑2 will potentially shift notoriety of OGN therapeutics further into public media platforms. Currently, there are 10 approved OGN therapeutics on the market (see Table 1).


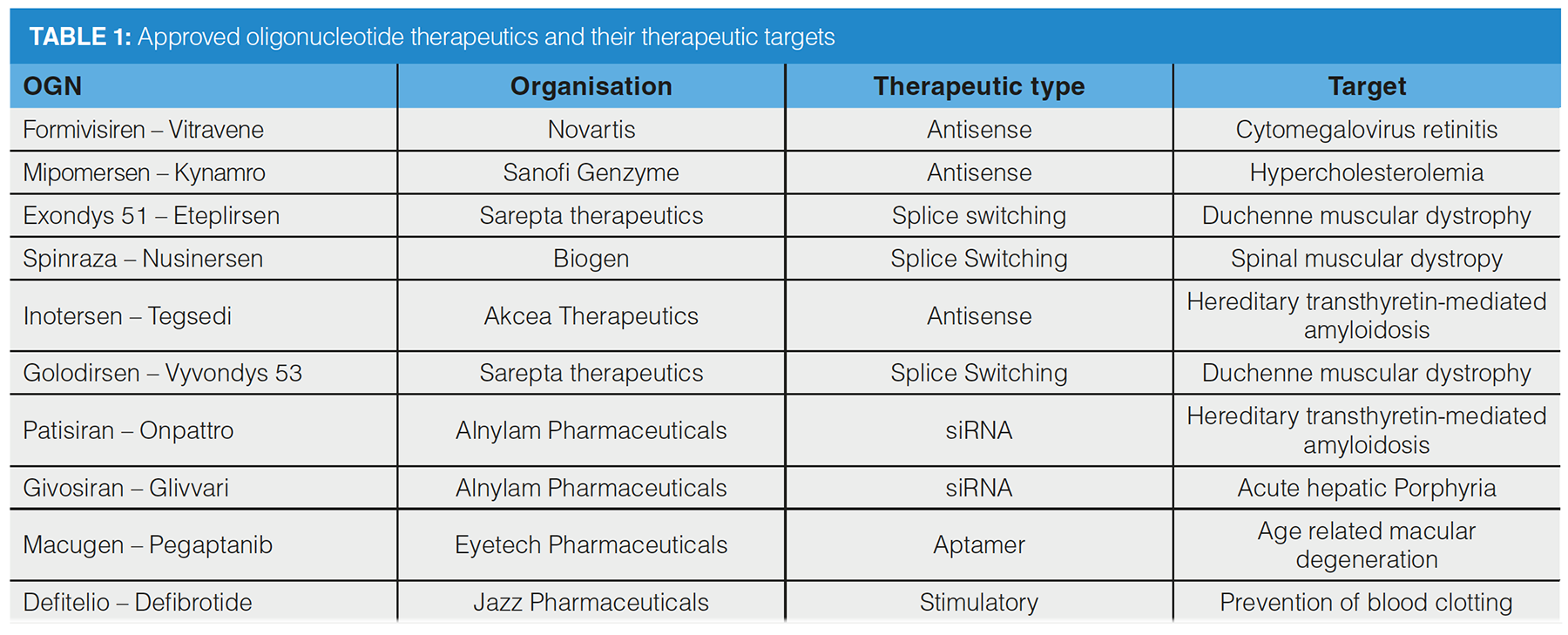
Among the types of OGN under this term are antisense oligonucleotides (ASOs) and splice‑switching oligonucleotides (SSOs), antagomirs, short interfering RNA (siRNA), aptamers, immunomodulatory OGNs, and messenger RNA (mRNA) (Figure 1). Initial development programmes observed that chemical modification led to increases in efficacy, resistance to degradative enzymes, and reduction in renal clearance rates. More recently, optimization of OGN delivery strategies has led to increased clinical success (1).

ASOs mediate the destruction of mRNA encoding “proteins of interest” by complementarily hybridizing with an mRNA sequence, which enables RNase H to bind to the ASO/mRNA complex and enzymatically digest the mRNA. SSOs interfere with splice‑switching events by sterically inhibiting the spliceosome when hybridized with the mRNA. This results in splice variants that lack aberrant exons contributing to disease states or proteins with homology to the healthy protein (2). Antagomirs (otherwise known as anti-miRs) also complementarily bind to their target, but these target micro-RNA (miRNA) molecules are involved with silencing of mRNA in disease states (3). ASOs, SSOs, and antagomirs are short, single-stranded OGNs ranging between around 19–22 nucleotides for ASOs or SSOs and 8–15 nucleotides for antagomirs. siRNA OGNs are RNA duplexes that also mediate mRNA degradation, but use a different process to ASOs. The two strands of the siRNA are named the passenger and guide strand, and are 20–24 nucleotides long. The guide strand is integrated into the cellular multiprotein RNA induced silencing complex (RISC), to enable sequence dependent degradation via hybridization with a target mRNA and sequential enzymatic digestion (4).
Increasing in size (approximately 50 nucleotides long) are aptamers; RNA-based OGNs that exhibit a 3D conformation and function to bind to ligands (5). These types of OGN therapeutics modulate the function of proteins of interest by sterically blocking receptor sites that are associated with disease or binding to free proteins. Aptamers can, in theory, be designed to bind to any target, much like antibodies. Immunomodulatory OGNs vary in size and structure to affect the immune response. Stimulation of the immune system using these molecules may co-function with other medical programmes, such as vaccination (6). The largest therapeutic OGNs are mRNA molecules that are used for in vivo translation of proteins of interest. mRNA therapeutics can code for proteins that are absent in disease, stimulate immune responses or act as viral protein encoding vaccines (7).
OGN Manufacturing and Impurity Profiles
Most small OGNs are synthesized by solid phase phosphoramidite chemistry using a cyclic process of detritylation, coupling, and capping nucleotides of the growing oligomer (8). Although each cycle efficiency is high (> 98%), as the target molecule grows in size; product yield reduces. The manufacturing batch consists of both the target OGN (the FLP or full‑length product) and OGN failed sequences, which are closely related OGNs, that is, aberrantly made because of failures in the synthesis process. Among the cohort of OGN-failed sequences are “shortmers”, “longmers”, diastereoisomers, depurinated OGNs, and OGNs that have not been fully deprotected, detritylated or chemically modified (9). Solid‑phase synthesis is unable to produce OGNs larger than 150 nucleotides and so larger mRNAs are produced by enzymatic in vitro transcription (IVT) (10). This is a process where RNA polymerase transcribes mRNA from a plasmid template encoding key structural elements, such as the 5’- and 3’- untranslated regions, the open reading frame, and the poly A tail. OGN impurities caused by IVT may be failed sequences (lacking structural elements), duplexes caused by mRNA self-complementarity, or originate from the DNA template used in the transcription process.
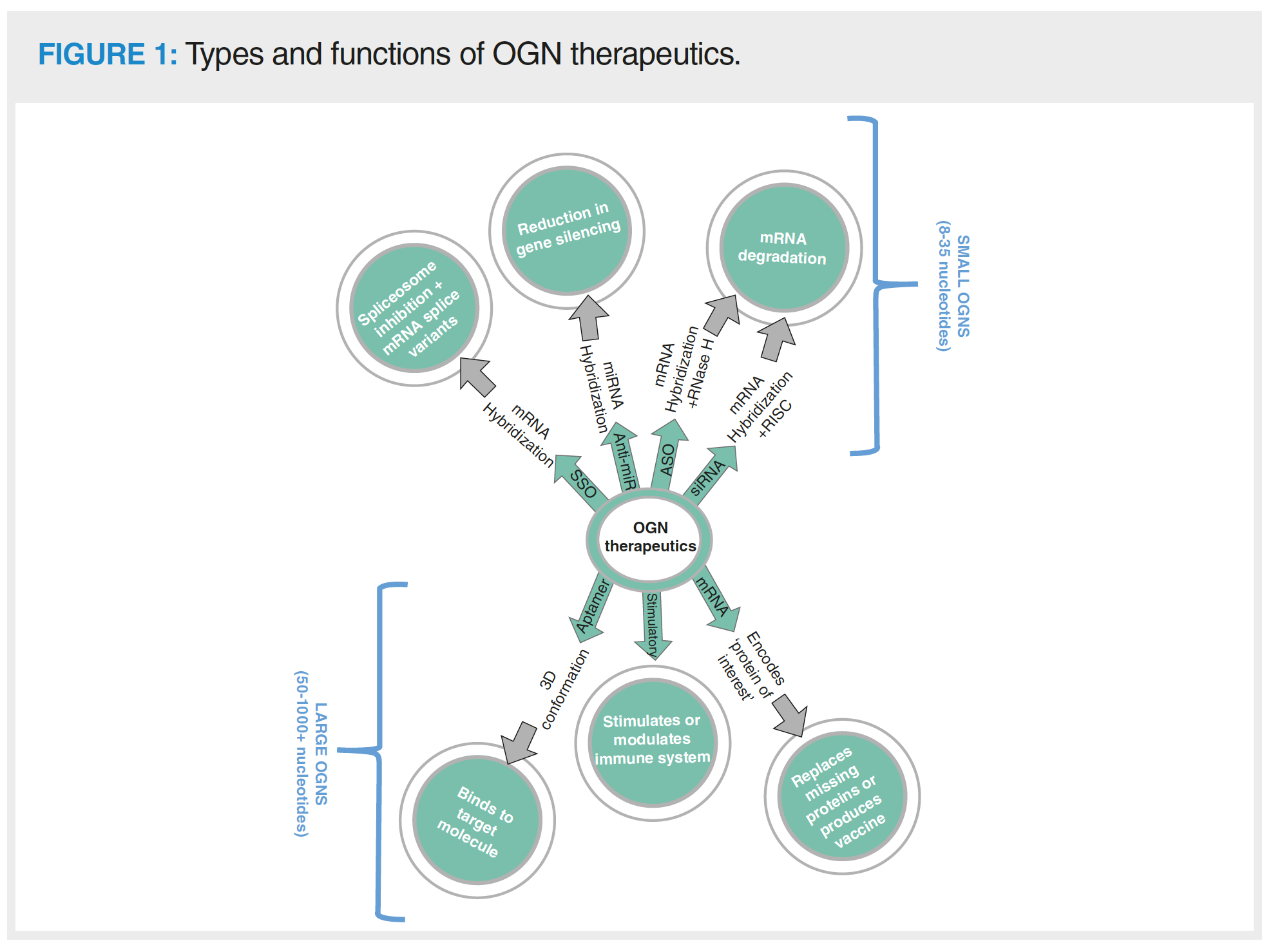
OGN therapeutics are commonly chemically modified to increase efficacy, reduce renal clearance rates, and increase their resistance to degradation by nucleases (1,11). There are multiple sites for modification, including the OGN bases, ribose sugar, and phosphate backbone (Figure 2). Different types of OGN therapeutic contain varying levels of chemical modification according to how well modifications are tolerated in relation to molecular function. ASOs are generally fully phosphorothioated as thioation of the phosphate backbone does not impede RNase H enzymatic activity but protects the OGN from nuclease degradation. Phosphorothioation is less well tolerated in other OGN types (such as siRNA) due to negative effects on OGN efficacy (1). Use of phosphorodiamidate morpholino SSOs is gaining popularity due to the ability to inhibit mRNA degradation, which allows steric blocking of the spliceosome for exon skipping (11).

Common modifications to the ribose of RNA nucleotides targets the 2’ carbon position, where the hydroxyl group is substituted for another moiety, such as a 2’-fluoro, 2’-O-methoxy or 2’-O methoxyethyl. Modifications to the ribose can be used to tune efficacy and enzymatic resistance. For example, siRNA with a combination of fluoro- and O-methoxy modifications demonstrates increased function and RISC loading efficiency (1). The ribose has a flexible conformation, which could be reduced by implementing locked nucleic acids (LNAs) into the OGN. LNAs constrain the movement of the ribose, which can assist to reduce off target interactions and associations, such as misloading of the siRNA passenger strand into the RISC. The LNA modification also demonstrates high binding affinity and is commonly integrated with other modification types, such as in LNA-2’-O-methyoxy-RNA phosphorohioated antagomirs (12).
Modifications to the bases can affect immunogenic response to the pharmaceutical (11). 5-methyl cytosine is commonly found in nature as an epigenetic modification. When found in OGN therapeutics, it increases duplex stability and reduces Toll-like receptor activation (thus reducing OGN immunogenicity). Synthetic analogues such as C5-thiazole or 5-methoxy uridine have also been developed for implementation into OGN therapeutics. Incorporation of analogue bases into mRNA may also increase the translation rate of the encoded protein of interest as a result of their stabilizing effect on the RNA structure (7).
In the last decade, OGN therapeutic clinical progress suffered a bottleneck effect because of challenges with drug delivery. Difficulties were observed in regards to delivering the OGN to the correct tissue or penetrating the target cell. This meant that high pharmaceutical doses had to be administered, causing immunogenic responses and toxicity, which led to further improvements to OGN medicinal chemistry via conjugation or encapsulation strategies (13). OGN therapeutics are often delivered within lipid nanoparticles or conjugated to cell delivery molecules (such as GalNac or cholesterol) that target specific cell types. Increasing chemical modification either within the OGN or with the addition of delivery vehicles increases the complexity of manufacturing batches in respect to potential OGN structures that are present.
Analytical Strategies for OGN Therapeutics
OGN therapeutics are not classified as either small molecules or biologics (14). However, they have characteristics that align with molecules in both categories. For example, like biologics, they contain complex chemistries but like small molecules, they are often made synthetically. Regulatory bodies and leading experts have published guidance on analytical strategies to characterize drug substance or product in respect to OGN identity, potency, quality, and purity (9,14,15). Analytical approaches contained within The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines are advised to be followed as much as possible. However, there is not a “one size fits all” methodology for reporting and monitoring critical quality attributes due to large differences in OGN structure and function. Standard methods for reporting typical attributes, such as residual solvents using gas chromatography are advised.
Analytical strategies for the characterization of OGN therapeutics are multifaceted to identify critical attributes, such as sequence, conformation, and structure. Chromatography sits firmly within the analytical strategy for post‑manufacture purification, characterization of impurities and structural variants, pharmacokinetic studies, analysis of drug formulations, and resolution of structures prior to mass spectrometry detection. Table 2 outlines liquid chromatography (LC) columns and recommended mobile phases for the analysis of OGNs.

High resolution liquid chromatography–mass spectrometry (LC–MS), size‑exclusion chromatography (SEC), and capillary gel electrophoresis (CGE) are used for analysis of molar mass, OGN size, and folding (16–18). These characteristics indicate OGN identity based upon retention time or mass. Differences in analytical retention time between manufacturing batches point towards changes to the OGN structure or conformation. CGE resolves OGNs by length and has been applied as a quantitative approach for purity analysis, although it requires a denaturant within the analysis buffer to linearize the OGN prior to the analysis. CGE is challenged by buffer effects, sampling bias, and cation adduction when coupled to mass spectrometry (MS) detection (19).
SEC may also be used to analyse OGN length, but can also point toward structural conformation when analysing molecules based upon their hydrodynamic radius (20). In relation to aptamers, it is especially important to characterize conformation as this is directly linked to ligand binding. SEC can also be used to understand the polydispersity of drug delivery conjugate molecules by analysing their hydrodynamic radii (14). SEC is challenged by low resolution and the chromatographer must optimize the pore size of the stationary phase, in relation to the size of the OGN, to achieve best resolution.
Further interrogation of structure is performed by analysing OGN sequence, which indicates why an OGN has an aberrant conformation or fails to hybridize accurately. MS interfaced with CGE or LC can identify potential mass differences between OGNs of smaller lengths (< 100 nucleotides) and be used for sequencing using a tandem approach (21). Electrospray ionization mass spectrometry (ESI-MS) is a particularly useful MS technique to interface with chromatographic separation as it preserves OGN structures thanks to a soft ionisation technique. The OGN is further fragmented using collision induced dissociation to produce predictable fragment patterns, as outlined by McLuckey and Habbibi‑Goudarzi in the 1990s (22). OGN LC–MS sequencing is challenged by increasing chemical modification of the OGN because this reduces the predictability of MS fragmentation patterns. MS analysis is also challenged by compatibility of chromatographic modes or mobile phases that reduce the efficiency of ionisation (23,24). An alternative method of LC–MS OGN sequencing is to chemically or biologically digest the OGN prior to MS analysis. Chemical and enzymatic digestion of an OGN sample produces fragments that are resolved in high resolution mass spectrometry (HRMS), and the OGN “ladder” is then sequenced by calculating the mass difference between the fragments (25,26). MS sequencing can identify erroneous sequences, which are crucial for function, such as the mRNA 5’-cap or siRNA failure sequences causing mismatches in duplex formation.
Chromatography plays a crucial role in the separation of OGN impurities from the full length product. These failed sequences can be structurally and chemically very similar to the FLP and often co-elute or exhibit low resolution from it (9). Failed sequences are resolved using a number of chromatographic modes, including string anion exchange (SAX), ion pair reversed phase (IP-RP), Hydrophilic Interaction chromatography (HILIC), and mixed mode chromatography (MMC). Commonly, industrial laboratories use IP-RP or SAX separation, depending on the target mechanism of separation.
Using SAX, OGN separation mechanisms are sequence dominant, which enables isomer and structural variant resolution (27). In addition to the ionic phosphate backbone, the OGN bases retain to the column in the order of adenine > thymine > cytosine > guanine at pH 8. Sequence dependent separation mechanisms may be further enhanced by increasing the mobile phase pH to ionize tautomeric guanine and thymine bases, which alters the retention order to adenine > cytosine > thymine > guanine (pH12) (28). OGNs with low levels of phosphorothioation (such as siRNA) are resolved using sodium chloride as the eluting salt. However, OGN elution from the column becomes more challenging as the level of phosphorothioation increases, leading to the requirement for a stronger displacer, such as sodium perchlorate. SAX is a useful tool to resolve OGN impurities that structurally differ from the FLP, such as linkage isomers that have undergone phosphoryl migration (29). In addition, the chromatographer can characterize diastereoisomer conformations in OGNs with a small number of chiral phosphorothioate bonds. As levels of phosphorothioation increase, diastereoisomer conformations increase in number, leading to broad peak shapes of fully phosphorothioated OGNs using SAX–LC.
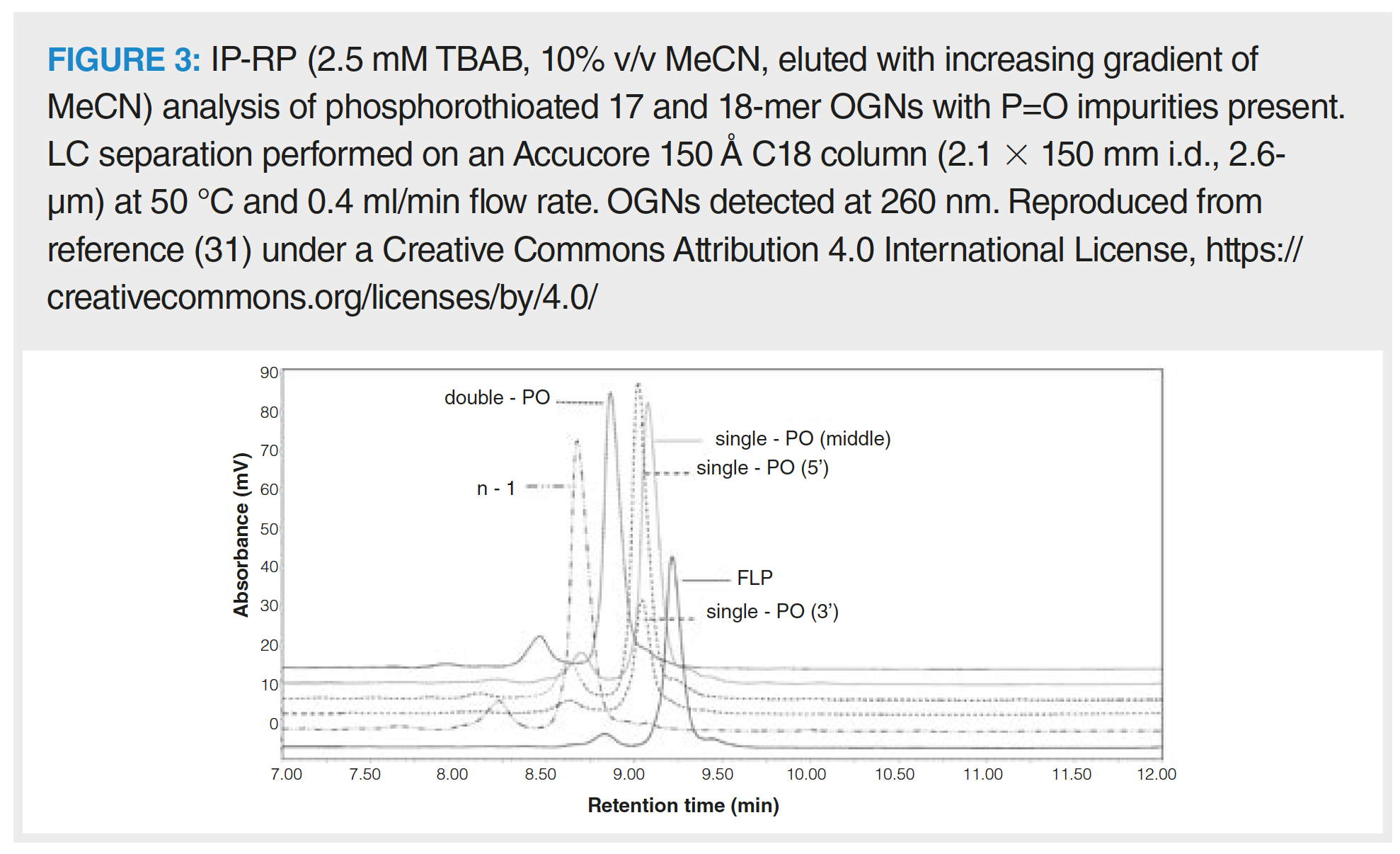
Solid phase synthesis of fully phosphorothioated OGNs creates P=O impurities: phosphorothioated OGNs containing phosphodiester bonds. These are only partially resolved from the FLP using SAX because of the electronegativity difference between sulphur and oxygen on the phosphate backbone (30). However, this is also achievable using IP-RP, as shown in Figure 3 where fully phosphorothioated OGNs and their phosphodiester impurities demonstrate differing retention behaviour (31). IP‑RP pairs the OGN with alkyl amines to neutralize the ionic charge imparted by the OGN backbone. Typical alkyl amine ion‑pair reagents (IPR) include triethylamine (TEA), diisopropylethylamine (DIPEA), tributylamine (TBA), diethylcylohexylamine (DMCHA), tetrabutylamine (TetBA) or hexylamine (HA) (32,33). The hydrophobic retention of OGNs is partly based upon OGN size as the IPR associates with each internucleotide bond. In addition, the OGN bases can hydrophobically interact with the reversed-phase stationary phase in the strength order of cytosine > guanine > adenine > thymine (although uracil is not as hydrophobic as thymine), creating a sequence‑based separation mechanism (34). More hydrophobic IPRs tend to partition more strongly toward the stationary phase, which creates a saturated layer that sterically inhibits secondary interactions between the OGN bases and the stationary phase. Size or sequence separations can be manipulated depending on the aim of separation by using more hydrophobic, linear IPRs (such as HA) for size dominance, or branched, less hydrophobic IPRs (such as TEA) for sequence dominance (35).

Lower flow rates have a demonstrably improved effect on OGN resolution in IP-RP, due to increased mass transfer effects (34). In addition, increased temperatures linearize OGN structures to further improve mass transfer. Analysis temperature can also be utilized to understand OGN duplex formation, which is useful for the analysis of siRNA OGNs. IP-RP analysis of duplex formation is performed by melting temperature (Tm) analysis to indicate mismatches in the duplex and OGN sequence isomerism. IP-RP applied at a range of temperatures indicates what temperature the duplex dissociates at. Changes to the OGN Tm indicate isomerism and mismatching within the duplex (36).
The majority of separations are conveniently achieved using methanol or acetonitrile as organic solvent. The use of IP-RP in conjunction with a strongly hydrophobic IPR enables the analysis of highly phosphorothioated OGNs because it reduces the resolution of diastereoisomers of identical size, which results in narrower chromatographic peaks. This effect enables shortmer/longmer OGN impurity analysis of OGNs with high levels of the modification, such as ASOs or SSOs. Challenges of higher limits of detection remain in IP-RP as a result of these broadened phosphorothioated OGN peak widths, which lead to higher reporting thresholds for OGN impurities (9).
IP-RP is a favoured choice for OGN separations because it is compatible with MS for impurity analysis, enabling a larger cohort of impurities to be characterized. The concentration of IPR within the mobile phase is usually decreased when coupling IP-RP with MS because high concentrations of IPR reduce MS ionisation efficiency. This may, in some cases, lead to negative impacts upon resolution of OGN impurities from the FLP. An addition of acidic alcohol hexafluoroisopropanol (HFIP) to the mobile phase can increase ionisation efficiency by increasing the surface charge on the ESI droplet (37). Using a combination of TEA and HFIP in the mobile phase has demonstrated the ability to resolve a range of OGN impurities, such as shortmers/longmers and P=O variants. A TEA:HFIP mobile phase is also suitable for phosphorothioated OGN analysis as it reduces diastereoisomer resolution and narrows OGN peak widths (38). HFIP forces the IPR to partition further toward the stationary phase, reducing secondary phase‑OGN base interactions and increasing size dominant separations. Resolution of one nucleotide is achieved using these conditions (39). The classical C18 column is a reliable starting point when analysing OGN therapeutics, but the chromatographer is advised to consider alternate, bonded phase chemistries. Phenyl-, amide- or cholesterol-bonded phases provide alternate selectivities and may be more appropriate for impurity analysis of chemically modified OGNs due to the provision of alternate types of interactions, such as pi-pi associations (40).
As the regulatory guidance on OGN therapeutic characterization is limited, orthogonal chromatographic techniques can provide enhanced selectivity and resolution (due to improvements in peak capacity). Examples of multidimensional chromatographic approaches for OGN therapeutics are limited, although demonstrate that a multitude of approaches can be taken. Various modes of LC can be combined (SAX, SEC, IP-RP, HILIC) and applied with both heart-cut and comprehensive separation approaches (41–44). Increases in 2D-LC popularity, owing to its orthogonal nature, suggests there are sure to be more multidimensional methods developed in the near future.
Conclusion
Chromatographic separations play critical roles within the multifaceted analytical strategy for OGN therapeutic characterization. There is a diverse range of OGN therapeutic types and various levels of chemical complexity imparted by a number of modifications. This leads to large diversity in analyte nature and interactions within chromatographic separations, which creates the requirement for multiple orthogonal analytical methods for OGNs. Analytical challenges associated with OGN therapeutic chromatography are related to increased chemical complexity, OGN size, and chemical similarity of OGN impurities to the FLP. Increasing chemical complexity, such as phosphorothioation or conjugation, can broaden peak widths and raise limits of detection and quantification. Increased modification also increases the likelihood of impurity and FLP co‑elution. As OGN length increases, resolution is harder to achieve, which may impact upon the chromatography of larger OGNs, such as mRNA or aptamers. OGN impurities are chemically and structurally very similar to the FLP and benefit from advances in chromatographic technology (such as higher efficiencies) and higher peak capacities. This helps purify the products of manufacturing, identify impurities, and analyse pharmaceutical quality attributes. Further developments to analytical methods in the coming years will hopefully overcome some of these analytical challenges and make OGN therapeutics even safer and publicly accepted as a result.
References
- A. Khvorova and J.K. Watts, Nature Biotech. 35(3), 238–248 (2017).
- A.M. Rossor, M.M. Reilly, and J.N. Sleigh, Pract. Neurol. 18(2), 126 (2018).
- J. Stenvang, A. Petri, M. Lindow, S. Obad, and S. Kauppinen, Silence 3(1), 1 (2012).
- B. Hu, Y. Weng, X.-H. Xia, X.-J. Liang, and Y. Huang, J. Gene Med. 21(7), e3097 (2019).
- S.M. Nimjee, R.R. White, R.C. Becker, and B.A. Sullenger, Ann. Rev. Pharma. Toxicol. 57, 61–79 (2017).
- P. Bodera, W. Stankiewicz, and J. Kocik, Pharmacol. Rep. 64, 1003–1010 (2012).
- O.V. Sergeeva, V.E. Koteliansky, and T.S. Zatsepin, Biochem. (Mosc.) 81(7), 709–722 (2016).
- S. Roy and M. Caruthers, Molecules 18(11), 14268–14284 (2013).
- D. Capaldi, A. Teasdale, S. Henry, N. Akhtar, C. Den Besten, S. Gao-Sheridan, M. Kretschmer, N. Sharpe, B. Andrews, B. Burm, and J. Foy, Nucleic Acid Ther. 27(6), 39–322 (2017).
- H. Kwon, M. Kim, Y. Seo, Y.S. Moon, H.J. Lee, K. Lee, and H. Lee, Biomaterials 156, 172–193 (2018).
- W. Brad Wan and P.P. Seth, J. Med. Chem. 59(21), 9645–9667 (2016).
- K. Lennox and M. Behlke, Pharma Research (AAPS) 27(9), 1788–1799 (2010).
- R.L. Juliano, Nucleic Acids Research 44(14), 6518–6548 (2016).
- D. Capaldi, K. Ackley, D. Brooks, J. Carmody, K. Draper, R. Kambhampati, M. Kretschmer, D. Levin, J. McArdle, B. Noll, R. Raghavachari, I. Roymoulik, B.P. Sharma, R. Thurmer, and F. Wincott, Ther. Innov. Regul. Sci. 46(5), 611–626 (2012).
- US Food and Drug Administration, CMC Regulatory Considerations for Oligonucleotide Drug Products: FDA Perspective (M. Sapru/FDA, Silver Spring, Maryland, USA, 2017). https://bit.ly/2RyIxzQ, accessed 16 September 2020.
- M. Madsen, S. Roussis, E. Schniepp, C. Rentel, and D. Capaldi, Rapid Commun. Mass Spectrom. 33(22), 1774–1780 (2019).
- E. Largy and J.-L. Mergny, Nucleic Acids Research 42(19), e149 (2014).
- K.E. Allan, C.E. Lenehan, D.A. Khodakov, H.J. Kobus, and A.V. Ellis, Electrophoresis 33, 1205–1214 (2012).
- J.V. Bonilla and G.S. Srivatsa, Handbook of Analysis of Oligonucleotides and Related Products (CRC Press, Boca Raton, Florida, 2011).
- R.R. Alieva, E.G. Zavyalova, V.N. Tashlitsky, and A.M. Kopylov, Mendeleev Communications 29(4), 424–425 (2019).
- M. Smith, Rapid Commun. Mass Spectrom. 25(4), 511–525 (2011).
- S.A. McLuckey and S. Habibi‑Goudarzi, J. Am. Chem. Soc. 115(25), 12085–12095 (1993).
- M.B. Beverly, Mass Specrom. Rev. 30(6), 979–998 (2011).
- S. Studzińska, Talanta 176, 329–343 (2018).
- J. Farand and M. Beverly, Anal.Chem. 80, 7414 (2008).
- H. Gao, Y. Liu, M. Rumley, H. Yuan, and B. Mao, Rapid Commun. Mass Spectrom. 23(21), 3423–3430 (2009).
- J.R. Thayer, V. Barreto, S. Rao, and C. Pohl, Anal. Biochem. 338(1), 39–47 (2005).
- J.R. Thayer, G. Gendeh, S. Rao, D. Jamieson, C.A. Pohl, and Y. Agroskin, “Performance Improvements for High Resolution Anion‑Exchange Oligonucleotide Separations Using Small Particle Substrates”, Thermo Scientific, USA (2013).
- J.R. Thayer, S. Rao, N. Puri, C.A. Burnett, and M. Young, Anal. Biochem. 361(1), 132–139 (2007).
- B. Bergot and G. Zon, Ann. N.Y. Acad. Sci. 660, 310–312 (1992).
- E.D. Close, A.O. Nwokeoji, D. Milton, K. Cook, D.M. Hindocha, E.C. Hook, H. Wood, and M.J. Dickman, J. Chromatogr. A. 1440, 135–144 (2016).
- L. Gong, Rapid Commun. Mass Spectrom. 29(24), 2402–2410 (2015).
- S.G. Roussis, M. Pearce, and C. Rentel, J. Chromatogr. A. 1594, 105–111 (2019).
- 3M. Gilar, K.J. Fountain, Y. Budman, U.D. Neue, K.R. Yardley, P.D. Rainville, R.J. Russell Ii, and J.C. Gebler, J. Chromatogr. A. 958(1–2), 167–182 (2002).
- S. Studzińska, R. Rola, and B. Buszewski, J. Pharm. Biomed. Anal. 138, 146–152 (2017).
- S.M. McCarthy, M. Gilar, and J. Gebler, Anal. Biochem. 390, 181–188 (2009).
- B. Chen, S. Mason, and M. Bartlett, J. Am. Soc. Mass Spectrom. 24(2), 257–264 (2013).
- N. Elzahar, N. Magdy, A. El-Kosasy, and M. Bartlett, Anal. Bioanal. Chem. 410(14), 3375–3384 (2018).
- J. Kim, B. Basiri, C. Hassan, C. Punt, E. van der Hage, C. den Besten, and M.G. Bartlett, Mol. Ther. Nucleic Acids. 17, 714–725 (2019).
- S. Studzińska, L. Pietrzak, and B. Buszewski, Chromatographia 77(23), 1589–1596 (2014).
- Q. Li, F. Lynen, J. Wang, H. Li, G. Xu and P. Sandra, J. Chromatogr. A. 1255, 237–243 (2012).
- P.W. Álvarez Porebski and F. Lynen, J. Chromatogr. A. 1336, 87–93 (2014).
- S.G. Roussis, I. Cedillo, and C. Rentel, Anal. Biochem. 556, 45–52 (2018).
- A. Goyon and K. Zhang, Anal. Chem. 92, 5944–5951 (2020).
Christina Jayne Vanhinsbergh is a researcher at the University of Sheffield, UK. Her Ph.D. research focused on the development of multidimensional chromatographic methods for the analysis of therapeutic oligonucleotides. Currently, she is researching DNA alkylation from environmental compounds using LCÐMS methodologies. Christina is the membership officer for the Chromatographic Society, UK.
Direct correspondence to: amatheson@mjhlifesciences.com













Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.


Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

