Profiles in Practice Series: Stewards of Drug Discovery — Developing and Maintaining the Future Drug Candidates
LCGC North America
process is interesting and involved, and in this month's installment of "MS - The Practical Art," Michael Balogh continues the Profiles in Practice series by exploring this topic with featured scientists Michele Kelly and Mark Kershaw.
Drug discovery generally is thought of as creation of a compound that possesses the potential to become a useful therapeutic. Successful discovery rests on a number of elements, one of which is early metabolite characterization of a novel chemical entity (NCE), which we addressed in the February 2005 installment of this series. Another element that supports discovery is analytical instruments and their ability to identify the NCE and purify it for storage and distribution downstream. This month's installment of "MS — The Practical Art" focuses on this latter aspect of discovery.



Michael P. Balogh
Today, pharmaceutical companies like Pfizer (Groton, Connecticut) and biotechnology companies like Neurogen (Branford, Connecticut) work toward a common goal: developing and maintaining proprietary libraries of registered compounds in standardized format for distribution and use. As Michele Kelly, associate director of analytical chemistry, Pfizer, told me, "We see ourselves as the stewards of Pfizer's compound library. We are involved at the beginning, when the compound is first registered, and we are responsible for 10 years or more — whenever the compound is distributed for use — for its purity, identity, concentration, and suitability."
The instrumental need imposed by pharmaceutical invention and development is much the same for any company, regardless of its size. How the need is met, however, varies. It depends upon available resources, the evolution of instrument technologies, and the abilities of those who operate the instruments. Over little more than a decade, a number of instrumental practices have changed significantly. Particularly in liquid chromatography–mass spectrometry (LC–MS), dramatic improvements in design and software have induced large shifts in pharmaceutical practices.


This Months Featured Scientists
The proliferation of MS as a tool for characterizing the earlier steps in drug discovery is, perhaps, the largest motivator of change in the pharmaceutical industry. The growth of automated synthesis, characterization, and archiving led to a demand for increasingly reliable instrumentation. Once available, this instrumentation produced greater confidence in the results as a basis for expected results and the creation of standard protocols.
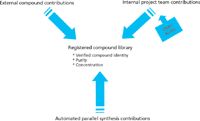
The advantages of automated synthesis are not restricted to analytical chemists. They also extend to medicinal chemists doing invention and to biologists, who are concerned with sample integrity as the basis for having made proper determinations of activity. Yet challenges remain for the stewards of library quality. These are illustrated in Figure 1, which shows the variety of contributions that a company must manage, regardless of source, to maintain a library effectively over the long term.

The Rise of Nonspecialist Systems for Initial Identity
Some farsighted pharmaceutical companies such as Pfizer engaged instrument manufacturers, notably those with more diverse capabilities, to create an interface by which nonspecialists could submit samples. Behind the scenes (a situation that reminds me of the movie Wizard of Oz, where the wizard stands behind a curtain working levers) highly trained mass spectrometrists monitored the systems' performance and calibration and diagnosed malfunctions as they arose.Frank Pullen (1,2) (Pfizer, Sandwich, U.K.) is credited with the earliest published work on automated, open-access configurations. In it, he relates his experience using MS software, which he adapted for use with diverse LC and GC inlets.
The early nonspecialist systems were simple flow-injection, isocratic systems — essentially an LC inlet connected to a single quadrupole mass spectrometer employing, predominantly, electrospray ionization. A submitter would log a sample identification and then leave the sample vial in a tray. Later, he or she would return for a spectrum, which appeared in the printer outbox. In short order, submitters were offered a few limited, general-purpose gradients. Automated e-mailing of results followed as the software increasingly became attuned to catching likely errors. Empty bottles or incorrectly concentrated samples would skew a result wildly. And although little could be done to correct such problems, the software could flag the affected result as an outlier. As LC–MS usage increased, sample backlogs reminiscent of traffic jams became notorious disrupters. But resolving the backlogs was easy, as one needed only to arrange a more suitable combination of instruments, location, capacity, and uptime.
Thanks to the now-obvious practicality of walk-up analysis, not to mention the cost savings inherent in instrument sharing, the open-access model for synthetic chemistry analysis is now de rigueur for all pharmaceutical companies. Today, dedicated automated systems routinely purify and analyze samples and manage archives. Unattended submission stations stand ready to accommodate ever more esoteric needs. For example, direct probe for electron ionization is used for sample compounds that do not respond to electrospray (ESI) or atmospheric chemical ionization (APCI) (3).
At Pfizer, after a decade of use, the open-access system remains the basis for simple, immediate, compound identity confirmation. The sole criterion for its use, says Kelly, is "the necessary turnaround time that allows the chemists to continue their work." At its Groton, Connecticut research site, Pfizer maintains 20 open-access systems. And though the company periodically reviews whether the systems are satisfying chemists' needs, few changes based upon the systems' reliability and throughput at peak usage have been indicated.
The practical aspects of open-access design and use are indeed important. Chemists who need answers are not apt to be impressed with a system's added capability unless it is just as easy as performing thin-layer chromatography. I highly recommend two publications on this topic. One is by Arthur Coddington (4), and the other is by Lawrence Mallis (5). The authors expand on column choices and the benefits to be gained from proper site selection. They also illustrate details such as how to cut PEEK tubing correctly, and they thoroughly explain the advantages of doing so.
Increasing sophistication of automated capabilities has removed the problem of establishing the identity of newly synthesized compounds from the forefront. In doing so, however, it has spawned new imperatives: standardizing data input and output and managing data for subsequent interpretation and distribution. Hence, the recent and visible rise of a new branch of data science: informatics. Entire companies such as NuGenesis (now part of Waters Corporation, Milford, Massachusetts) were formed to respond to the new need, and software like AutoLynx, a MassLynx application manager (Waters), was invented.
Neurogen
Mark Kershaw is charged with devising analytical support for traditional medicinal chemists and the Neurogen High Speed Synthesis (HSS) team. The HSS team uses parallel solution phase syntheses to support discovery projects through rapid creation of chemical libraries. Key to achieving objectives is integrating novel workstations with the informatics platform to reduce resource demands and permit efficient movement of samples and data. Thus, the ability to interpret the established platform is a criterion for any methodology or technology that Kershaw's team adopts. In its characterization process, Neurogen uses MassLynx software (Micromass), which permits third-party programmers access to various input–output data and control "hooks."
Jeffrey Noonan, associate director of the HSS group, describes his company as technologically adept, a "mobile entrepreneurial epicenter of technology development." Neurogen's success in devising innovative ways to support drug discovery back that claim: just seven chemists can synthesize, purify, quantify, and characterize more than 100,000 compounds a year.
As with the original open-access systems, LC–MS data are viewed at the scientist's desktop via intranet web pages. Neurogen's platform supports QC functions: batch testing of incoming raw materials, tracking of library samples through the HSS workflow (including MS-directed purification), fraction management, and re-analysis (when required). Having met QC standards, established library compounds are distributed for use in biology screening and pharmaceutics profiling.
At Neurogen, purity is achieved primarily via automated parallel solid-phase extraction (SPE), which can process 352 library compounds in a 3-h unattended run. Combining liquid–liquid extraction with appropriate SPE media (silica or SCX) provides an effective means of removing common library impurities and reagents. Postpurification, LC–MS purity assessments are based upon automated evaluation of chromatographic peak and spectral data. Rules for purity have been developed, including such aspects as isomer detection and identification of starting materials and reagents. Suspect samples are flagged for MS-directed purification when the more cost-effective SPE methods fail, or about 10% of the time (6).
Few companies of any size would jettison gainfully employed equipment in favor of the next new thing on the market. Faced with a need to characterize an increasingly large number of library samples a few years ago, one could select a multiplexed LC–MS inlet system or several redundant LC–MS systems. Kershaw opted for the latter, installing two readily available time-of-flight (TOF) instruments (Waters LCTs) coupled with monolithic columns (50 mm × 4.6 mm Merck Chromalith), which could in relatively short order process the 100,000-plus samples in the library. High flow rates and short reverse gradients deliver 2-min run times that result in peaks 1–2 s wide. Unlike older quadrupoles, which could not scan fast enough for such narrow peaks, TOF instruments are effective because the target mass was well characterized. Very clean samples at low back pressure also gave column lifetimes of 20,000 injections.
High-throughput profiling capabilities currently are being developed. Although in the future, pharmaceutics characterizations will be performed as they are today, broad early indications of parameters like solubility are a useful feature of web-based informatics systems. When the ACQUITY ultrahigh pressure chromatography system (Waters) was introduced last year, Kershaw obtained one coupled with a Waters ZQ mass spectrometer, a quadrupole instrument that can scan to 5000 amu/s. Adding the new system increased specificity, spectral clarity, and sensitivity gains while not deviating from the established system workflow design.
Pfizer
Pfizer Central Research has developed an enviable track record for automation and production. One obvious and extensive change in its practice is the harmonization of methods over a number of worldwide research sites, including the company's contract contributors. Not long ago, dry samples were standard in many libraries, and they required unique stability testing and manipulation for distribution. So much handling invited human error, which manifested itself in subsequent screens and elsewhere. Today, all samples are maintained in dimethyl sulfoxide stock solutions, and they satisfy criteria for identity, purity (as determined by UV and evaporative light-scattering detection [ELSD]), and concentration.
Still, the concern that we must learn more about concentration is legitimate. The measurement associated with a compound's registration (that is, when it enters the library at "time 0") is critical to making accurate subsequent assessments. After "time 0" determinations and storage in dimethyl sulfoxide, changes in concentration affect calculations and the results of screens used to determine the value of the compound as a potential therapeutic. Drug discovery has adopted dimethyl sulfoxide as its reagent of choice. Although hygroscopic, dimethyl sulfoxide's viscosity and other properties make it the reagent of choice for storing and dispensing NCEs. And yet, despite being almost a universal solvent, dimethyl sulfoxide does not work well for some products. For instance, drying some less soluble compounds can leave a difficult-to-redissolve pellet in the vessel. Storage, usage, freeze-and-thaw cycles (water absorption issues), and various computational implications are all subjects of an ongoing debate reported by the Laboratory and Robotics Interest Group (www.lab-robotics.org).
Because both open access and purification standards are specified, Kelly's primary interest today is improving concentration assessments and better understanding end-use effects. Of particular concern is how samples are affected by their handling. Does the person accepting the sample store it during use at room temperature, or does he or she refrigerate it? What are the benefits of single-use containers versus multiple-use containers? At Pfizer, initial (first-registration) measurements of purity and suitability are made using UV and ELSD. But as with all areas of practice, where only limited information on detector response is available (for example, mass balance estimations for assessing degradation of a compound), no true universal method is available. However, the industry is eyeing two contenders for a universal method: ELSD and chemiluminescent nitrogen detection (CLND).
Gary Schulte (Pfizer), whom I encountered at last August's CoSMoS conference (www.cosmoscience.org), has monitored the ELSD–CLND debate for years. Here is what he had to say about it:
Quantitation methods for unknowns not using specific standards with their calibration curves is a hot topic on the discovery side of most pharmaceutical companies at the moment. It pits ELSD versus CLND methods in the LC–SFC world, and these methods versus NMR methods. The reason is that many folks are trying to read the concentration of their screening samples in a high-throughput mode.
At their current state of development, both ELSD and CLND instruments, although integrated in automated, unattended systems, are in need of improvement.
Pfizer currently favors the ELSD method, which, though more broadly useful, can incur a 20–30% quantitative error. Other means of adjusting concentration errors have been reported. Burke uses the scatter of results likely due to concentration effects from initial returns on IC50 screens to back-correct CLND results (7). Published work on ELSD using standards shows an average error of 8.3% (8). I.G. Burke's application uses no standards but adds the step of back-correcting the initial results. Anecdotal evidence suggests either detector, in the hands of an experienced practitioner, is a capable device. But neither has established a clear reputation for ruggedness in use, nor can either claim an extensive comparative base with more common detection such as UV and MS.
The drug discovery concept has changed significantly in less than a decade. In a hybridization of the traditional practice, project-oriented medicinal chemists who rely on automated processes still can purify their products via flash chromatography, although automated support processes play a much larger role today. Automation itself has matured into a more homogeneous, extensively used tool, whereas its successful use a decade ago was restricted to but a few isolated roles. The open-access concept, alive and thriving, has become a highly useful means of characterizing NCEs early in the discovery process. Subsequent tiers of quality control analysis offer a view of sample integrity at the baseline of initial dissolution, a baseline that will be monitored over the sample's lifetime.
References
(1) D.V. Bowen, M. Dalton, F.S. Pullen, G.L. Perkins, and D.S. Richards,
Rapid Commun. Mass Spectrom.
8
(8), 632–636 (1994).
(2) F.S. Pullen, G.L. Perkins, K.I. Burton, R.S. Ware, and J.P. Kiplinger, J. Am. Soc. Mass. Spectrom. 6(5), 394–399 (1995).
(3) L.O. Hargiss, M.J. Hayward, R. Klein, and J.J. Manura,http://www.sisweb.com/referenc/applnote/app-90.htm Novartis Pharmaceuticals, Summit, New Jersey, Sept. 2000.
(4) A. Coddington, J. Van Antwerp, and H. Ramjit, J. Liq. Chromatogr. Rel. Tech. 26(17), 2839–2859 (2003).
(5) L.M. Mallis , A.B. Sarkahian, J.M. Kulishoff, Jr., and W.L. Watts Jr., J. Mass Spectrom. 23(9), 889–896 (2002).
(6) J.W. Noonan, J. Brown, M.T. Kershaw, R.M. Lew, C.J. Lombardi, C.J. Mach, K.R. Shaw, S.R. Sogge, and S. Virdee, J. Assoc. Lab. Auto. 8, 65–71 (2003).
(7) I.G. Popa-Burke, O.Issakova, J.D. Arroway, P. Bernasconi, M. Chen, L. Coudurier, S. Galasinski, A.P. Jadhav, W.P. Janzen, D. Lagasca, D. Liu, R.S. Lewis, R.P. Mohney, N. Sepetov, D.A Sparkman, and C.N. Hodge, Anal. Chem. 76(24), 7278–7287 (2004).
(8) B.T. Mathews, P.D. Higginson, R. Lyons, J.C. Mitchell, N.W. Sach, M.J. Snowden, M.R. Taylor, and A.G. Wright, Chromatographia 60(11/12), 625–633 (2004).
Michael P. Balogh "MS — The Practical Art " Editor Michael P. Balogh is principal scientist, LC–MS technology development, at Waters Corp. (Milford, Massachusetts); an adjunct professor and visiting scientist at Roger Williams University (Bristol, Rhode Island); and a member of LCGC's editorial advisory board. Direct correspondence about this column to "MS— The Practical Art,"LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgcmag.com.


















Separating Impurities from Oligonucleotides Using Supercritical Fluid Chromatography
February 21st 2025Supercritical fluid chromatography (SFC) has been optimized for the analysis of 5-, 10-, 15-, and 18-mer oligonucleotides (ONs) and evaluated for its effectiveness in separating impurities from ONs.

