Control of Matrix Effects in Bioanalytical MS–MS Using On-line Multidimensional Solid-Phase Extraction
LCGC North America
The authors discuss a solid-phase extraction (SPE) clean-up procedure for drugs present in biofluids.
In the pharmaceutical industry, liquid chromatography–tandem mass spectrometry (LC–MS-MS) is already an established method for quality control and quantification of drugs in different matrices. Additionally, this hyphenated analytical tool is becoming more and more important in clinical chemical analysis (that is, in therapeutic drug monitoring).
LC–MS-MS is a very powerful analytical technique because it combines the separation power of LC with the sensitivity and selectivity of MS. However, LC–MS-MS possesses two major drawbacks when analyzing drugs in biological fluids. The first, associated with the mass spectrometer, is that the electrospray source is very susceptible to matrix-related ion-suppression effects. The second drawback concerns the LC. Because of irreversible adsorption and precipitation effects caused by high molecular weight sample components (for example, proteins) complex biofluids cannot be injected and analyzed directly.
Consequently, the analysis of complex biological fluids generally requires pretreatment steps aimed at the removal of unwanted matrix constituents from the sample. In bioanalytical LC, solid-phase extraction (SPE) is the predominant clean-up technique. However, the high matrix load of complex biofluids affects the efficiency of this extraction technique and gives rise to co-elution of interfering substances. This is particularly true for proteins, because many commercially available SPE sorbents are not biocompatible and cause nonspecific adsorption and/or precipitation of proteins. With on-line SPE, this in turn causes a clogging of the SPE column and shortens its lifetime dramatically. As a result, most sample clean-up procedures include a protein precipitation step in order to prevent these effects. However, protein precipitation cannot be automated easily and, in fact, Polson and colleagues (1) showed that protein precipitation is not efficient enough to remove all substances that cause ion suppression. In addition, protein-bound analytes are often co-precipitated. Thus, optimization of sample pretreatment should be directed toward improved efficiency–selectivity and automation. We have accomplished this with the development of a multidimensional SPE platform for extractive on-line clean-up and subsequent LC–MS-MS analysis of drugs with basic properties and low polarity in raw biofluids (Figure 1).


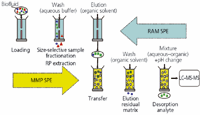
Figure 1: Schematic of the MD-SPE operational procedure. Analyte 5 squares, high molecular weight matrix component 5 triangles, low molecular weight matrix component 5 circles.
Experimental
Instrumental set-up formulations:
Figure 2 illustrates the multidimensional on-line SPE–LC platform. The system consists of three conventional HPLC pumps, two six-port switching valves controlled by an HTC PAL autosampler (CTC Analytics, Zwingen, Switzerland), a diode array detector (Merck KGaA, Darmstadt, Germany), a restricted access material (RAM) SPE column (5 × 4 mm i.d.) packed with LiChrospher
®
ADS RP18 material (Merck KGaA, Darmstadt, Germany), a mixed mode polymer (MMP) SPE column (20 × 1 mm i.d., research prototype provided by Waters Corporation, Milford, Massachusetts), and an analytical column (Zorbax RX-C18, 3.5 µm, 75 × 4.6 mm i.d., Agilent Technologies, Waldbronn, Germany).

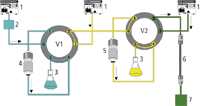
Figure 2: Schematic of the MD-SPE-LC system. 1 5 pumps, 2 5 autosampler, 3 5 waste, 4 5 RAM column, 5 5 MMP column, 6 5 analytical column, 7 5 detector, V1 and V2 5 valves.
A triple quadrupole MS (Quattro micro, Waters) equipped with an electrospray source was used for MS detection. The spectra were recorded in multiple reaction monitoring (MRM) mode. The mass transitions of the tricyclic antidepressants used as model drugs are as follows: amitriptyline (278.4 > 91.2), clomipramine (315.4 > 86), desipramine (267.4 > 71.9), doxepin (280.4 > 107.3), imipramine (281.4 > 86), maprotiline (278.4 > 250.3), nortriptyline (264.4 > 191), protriptyline (264.4 > 91.2), and trimipramine (295.5 > 104). The dwell time for each of these transitions was 0.25 s. Source temperature was set to 130 °C, desolvation temperature to 250 °C, desolvation gas flow to 700 L/h, capillary voltage to 3.5 kV, and cone voltage to 20 V.
For diode array detection, the instrumental set-up was slightly modified. The size of the RAM SPE column was 20 × 4 mm i.d. Fractionation on the RAM SPE column was performed at a flow-rate of 1 mL/min for 8 min and on the MMP SPE column at the same flow-rate for 6 min.
MD-SPE Procedure for Plasma
First, the raw biofluid (e.g., 20 μL) is injected directly onto the RAM SPE column. Because of the pore diameter (6 nm) of the sorbent, high molecular weight sample components larger than 15 kDa are size excluded and washed completely to waste within 2 min at a flow rate of 4 mL/min using an eluent composed of water/acetonitrile (95/5, v/v). Thereafter, valve 1 (V1) is switched, and the extracted low molecular weight sample components are eluted at a flow rate of 3 mL/min within 2 min onto the MMP SPE column using methanol as mobile phase. After this, chemoselective fractionation step valve 2 (V2) is switched and the target analytes are transferred at a flow rate of 0.8 mL/min onto the analytical column with an eluent composed of acetonitrile–5 mM ammonium acetate and 0.1% formic acid (40/60, v/v).
When applying only the RAM SPE column for sample clean-up, the mobile phase consisted of methanol/2 mM ammonium acetate (80/20, v/v) and the flow-rate was set to 0.8 mL/min.
Identifying Ion Suppression
To investigate the ability and the efficiency of the MD-SPE platform to reduce matrix effects, the experiments shown in Figures 1 and 2 were performed.
Monitoring of ion suppression effects was achieved by postcolumn infusion experiments. For that purpose, a T-piece was placed between the outlet of the LC and the MS source. A standard solution of the target analytes was then infused via the T-piece at a flow rate of 10 μL/min into the eluent stream and the MRM transitions (TIC profile) of the analytes were recorded by the MS (Figure 3).


Figure 3: Comparison of conventional RAM SPE and MD-SPE. Chromatograms of 100 õL each of plasma (a,c) and water (b,d) were recorded by a diode array detector at 220-400 nm. Samples (a) and (b) were analysed by applying a conventional RAM SPE procedure. Samples (c) and (d) were pretreated using the MD-SPE platform.
In addition, the MD-SPE platform was compared with a column-switching method that used only the RAM SPE column. For that purpose, a diode array detector was used to monitor the remaining matrix components after the reversed-phase (RP)–SPE clean-up step by scanning the wavelengths from 220 nm to 400 nm. As a reference sample, double-distilled water was used (Figure 4).

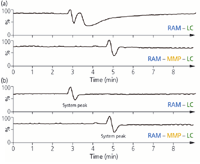
Figure 4: Comparison of conventional RAM SPE and MD-SPE. Infusion chromatograms were recorded by analysing 100 õL blank samples each of (a) plasma and (b) water. The chromatograms represent the TIC profile of all nine tricyclic antidepressants investigated.
To monitor the selectivity of the MD-SPE clean-up procedure, an MS scan (m/z 100-1500) of the same blank sample of human plasma was recorded (Figure 5). Finally, a human plasma sample (20 μL) was analyzed using the MD-SPE platform (Figure 6).

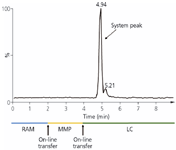
Figure 5: MS scan (m/z 100-1500) of an LC elution profile obtained after on-line MD-SPE of 100 õL human plasma (blank).
Results and Discussion
The separation power of the described MD-SPE platform relies on four chromatographic modes, that is, four separation dimensions. Each fulfils the criterion of orthogonality, meaning that they are independent of each other, which is a prerequisite for multidimensional separation (2).

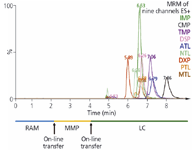
Figure 6: On-line MD-SPE-LCâMS/MS chromatogram of human plasma (20 õL) spiked with 30 ng/mL of each of the antidepressants investigated.
On the RAM SPE column (Figure 1), two chromatographic principles are applied simultaneously, namely, size-exclusion (SEC) and reversed-phase chromatography (RPC) (3). These represent the first and second dimension of the MD-SPE platform. When using the RAM packing, the size-exclusion corresponds to a molecular weight cut-off of approximately 15 kDa. For RPC, either a C4, C8, or C18 modification can be chosen. When fractionating and extracting a biofluid such as human plasma, all matrix components larger than 15 kDa are quantitatively eluted to waste, whereas a fraction of smaller components to which the target analytes belong is retained because of reversed-phase partitioning.
According to the MD-SPE procedure, this fraction is then eluted onto the MMP SPE column (Figures 1 and 2). Relating to the second criterion for multidimensionality, namely, comprehensiveness, the described method is fully comprehensive with regard to the extracted low molecular weight fraction, that is, the target analytes.
On the MMP SPE column, two additional chromatographic principles are applied simultaneously, namely, ion-exchange (IEC) and hydrophobic interaction chromatography (HIC).
These represent the third and fourth chromatographic dimensions of the MD-SPE platform. We applied a weak cation-exchanger to be able to extract and desorb drugs possessing basic properties by varying the pH value of the mobile phase accordingly.
The combination of SEC–RPC with IEC–HIC proves to be highly efficient for removal of MS-interfering high and low molecular weight sample components. This is documented in Figures 3–5.
Diode array detection revealed that SPE of a human plasma sample based solely upon RP-RAM extracts many low molecular weight plasma components. This is expected and results from the reversed-phase partitioning of plasma constituents possessing low polarity (Figure 3a).
However, when applying the combination of RAM and MMP SPE most of the remaining and potentially MS–interfering matrix components are eliminated (Figure 3c).
The postcolumn infusion chromatograms shown in Figure 4 demonstrate that the described MD-SPE platform for MS indeed eliminates ion suppression with regard to the analytes and matrix investigated. The only disturbance seen in the baseline is caused by valve-switching as confirmed by injecting a sample of double-distilled water.
Furthermore, we investigated blank plasma samples using an MS scan mode to prove the selectivity of the MD-SPE platform. A representative chromatogram shown in Figure 5 reveals that after MD-SPE, no compounds with the potential to cause ion suppression are detected. Thus, this experiment also conclusively shows that the described MD-SPE procedure provides an MS–adequate clean-up of biofluids.
Finally, human plasma samples were spiked with the model analytes; that is, nine different tricyclic antidepressants, and analyzed as per the described procedure. As shown in Figure 6 all of these drugs can be quantified without any disturbances related to LC or MS.
Conclusions
We have developed a novel, fully automated MD-SPE platform that allows the direct injection and integrated clean-up of biological fluids such as human plasma, as well as a highly selective extraction of defined target analytes. The method has a generic application potential with respect to basic analytes with pka values higher than 6.5 and moderate-to-low polarities.
The described MD-SPE platform proves to be efficient for the removal of MS–interfering matrix components and, therefore, permits an undisturbed LC–MS–MS analysis of highly complex samples.
Acknowledgements
The authors would like to thank Merck KGaA (Darmstadt, Germany) and Waters Corporation, (Milford, Massachusetts) for providing them with special SPE packings. They also would like to thank Roche Diagnostics GmbH (Penzberg, Germany) and CTC Analytics (Zwingen, Switzerland) for financial support and instrumentation.
References
(1) C. Polson et al.,
J. Chromatogr., B
785,
263-275 (2003).
(2) P. Schoenmakers, P. Marriott, and J. Beens, LCGC Eur., 335-339 (2003).
(3) K.-S. Boos and C.-H. Grimm, Trends Anal. Chem. 18, 175-180 (1999).
Karl-Siegfried Boos is Professor of Clinical Chemistry and Head of the Laboratory of BioSeparation at the Institute of Clinical Chemistry, University Hospital Grosshadern, Munich, Germany. Since 2000 he has been co-editor of Chromatographia. He can be reached by e-mail at boos@med.uni-muenchen.de.
Katrin Georgi studied chemistry at the University of Leipzig, Germany. She is currently a PhD student working on bioanalytical mass spectrometry under the supervision of Karl-Siegfried Boos.
















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).

Silvia Radenkovic on Building Connections in the Scientific Community
April 11th 2025In the second part of our conversation with Silvia Radenkovic, she shares insights into her involvement in scientific organizations and offers advice for young scientists looking to engage more in scientific organizations.


