Practical Considerations for Capillary Electrophoresis–Mass Spectrometry for Analysis of Biotherapeutics
LCGC North America


Coupling CE with MS presents some challenges. Here, we discuss the advantages of CE–MS over LC–MS, and the parameters that are important to obtain a stable CE–MS profile.
Biotherapeutics are a growing class of drugs that need extensive characterization of heterogeneities that are acquired during their manufacturing, storage, and distribution. Liquid chromatography–mass spectrometry (LC–MS) is widely considered a gold standard for characterization of biopharmaceuticals. Capillary electrophoresis (CE) offers a promising alternative to high performance liquid chromatography (HPLC), but one of the challenges that has often been cited is the difficulty in coupling it with MS. In this article, we discuss the coupling of CE with MS, advantages of CE–MS over LC–MS, some recent applications of the technology in the field of biotherapeutics, and the various parameters that are important to obtain a stable CE–MS profile.
Biotherapeutics are drugs based on proteins and nucleic acids that are expressed in living organisms using recombinant DNA technology (1). The first recombinant protein biotherapeutic was approved in 1982, and since then there has been tremendous expansion in the therapeutic protein market (2). With more than 100 biopharmaceuticals approved from 2014 to 2018, and many more in the pipeline, the industry has shown significant and consistent growth. Also, as the patents of many of the blockbuster biotherapeutics expire, development of biosimilars has been increasingly in focus within the past decade (3). Biosimilars are copies of the approved innovator biotherapeutic products that have been demonstrated to show equivalent safety and efficacy (4).
Biotherapeutics are different from chemically synthesized drug molecules, because they are produced in biological systems, and this leads to inherent heterogeneities in the products. For example, the generation of amino acid sequence variants during the production of a protein therapeutic, due to genomic mutations, nutrient starvation during clone selection, and translational errors, is quite common (2). Along with the use of biological hosts for production, the raw materials, manufacturing, and storage procedures may also alter properties of the product, including the physical and chemical properties, as well as cause denaturation of the product. These variations can change the biological activity of the product and affect its immunogenicity, thus affecting the safety and efficacy of the drug. It is therefore critical to perform extensive characterization of the product throughout the lifecycle of the product, from manufacturing to market (2).
Many orthogonal, high resolution techniques have been used for biological, immunological, and physiochemical characterization of biotherapeutics. Size-exclusion chromatography (SEC), nuclear magnetic resonance (NMR), liquid chromatography (LC), and capillary electrophoresis (CE) comprise, in part, the analytical toolbox for biotherapeutic characterization. A list of available tools for characterization of biotherapeutics is presented in Table I. Of these, LC coupled to mass spectrometry (MS) has become state-of-the-art technology for routine characterization of biotherapeutics in the industry (5). LC–MS, although widely accepted, suffers from several limitations for biopharmaceutical characterization, such as the requirement of relatively large amounts of sample (micrograms), limited performance in the analysis of post-translational modifications, and difficulties associated with detection of co-eluted peptides, usually due to ion suppression (6,7).


CE offers an alternative to LC, because it involves separation of analytes based on their different mobility under an electric field in narrow bore capillaries. This orthogonal mode of separation in CE, when coupled to highly sensitive MS detectors, makes it a complementary technique to LC–MS, and it is increasingly being applied for biopharmaceutical characterization (5,8,9). CE is available in various modes, such as capillary zone electrophoresis (CZE), capillary gel electrophoresis (CGE), capillary isoelectric focusing (cIEF), and micellar electrokinetic chromatography (MEKC), which allow for characterization of different attributes, such as the intact and reduced mass of a protein, charge variants, peptide mapping, and glycosylation patterns (5,10).
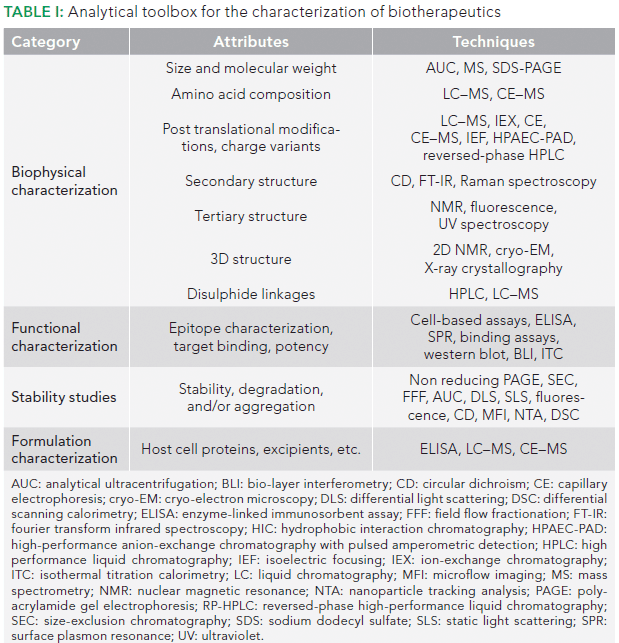
Unfortunately, the coupling of CE with MS can be challenging. Application of an electric field between CE capillary exit and MS inlet, while maintaining the electric field (to drive the CE separation and obtain electrospray, at the same time) is a major hurdle. The fact that the CE current is more than a hundred times larger than the electrospray current necessitates the development of adequate, safe electric circuits for handling and protecting the MS instrument. The meager flow rate in CE and the incompatibility of most CE buffers with MS are other hurdles in coupling CE with MS. It thus took quite a bit of time to develop an interface that allows this coupling. The currently available modes for CE–MS coupling can be broadly divided into two categories, namely sheath flow and sheathless interfaces, depending upon the use or lack of use of sheath fluid to establish the electrical contact between the two systems (11,12). Although the use of a sheath flow interface leads to dilution of the CE effluent that may reduce sensitivity, it also gives one an opportunity to manipulate the separated analytes chemically. For example, a higher pH of sheath fluid has been shown to generate only singly and doubly charged molecular ions. The sheath liquid composition can thus be altered to optimize MS detection without affecting CE separation (13). Sheathless interfaces have higher sensitivity, but have less stability. Sheath flow interfaces are preferred, but applications of CE coupled to either kind of interface have been reported (12). The coupling of CE can be done with both types of traditional MS, such as matrix-assisted laser desorption–ionization (MALDI), as well as electrospray ionization (ESI) type. Although MALDI coupling is simple, as compared to ESI–MS, the latter is preferred for CE coupling because the MALDI approach results in loss of resolution, signal suppression, and signal variability, as a result of matrix effects (14). A typical coupling of CE–ESI-MS with a sheath flow interface is depicted in Figure 1.

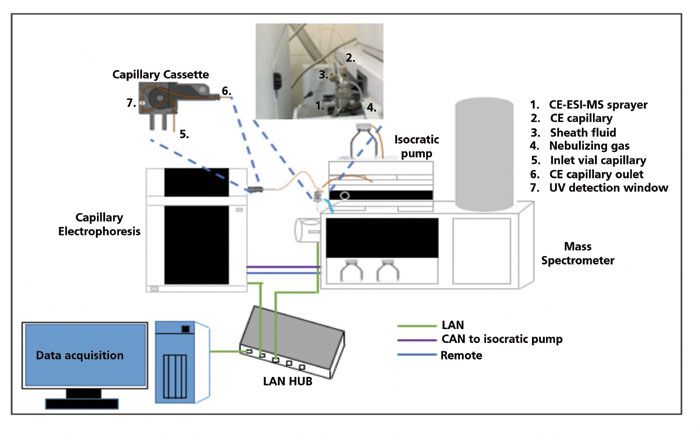
Figure 1: Schematic diagram representing CE-MS coupling.
CE–MS in the Biopharmaceutical Industry
CE is effective in the characterization of biotherapeutics, and it has been accepted by the U.S. Food and Drug Administration (FDA) for process development, characterization, and quality control activities. Researchers have reported 100% sequence coverage with CE–MS of monoclonal antibodies (mAbs), whereas LC–MS of the same sample missed several short peptides (2). CE–MS has also been shown to be useful for N-glycan and charge variant analysis of monoclonal antibodies (5). CE–MS has also been used for studies of site-specific mapping and multiple deamidations in peptides (15), host cell impurity characterization in mAbs (16), and post-translational modifications in peptides (7), among many other applications.
Comparison of LC–MS with CE–MS
CE does not use a stationary phase, and, as a result, there is a reduced probability of irreversible peptide loss in CE compared to LC (6). As compared to LC, CE offers the ability to work at very low flow rates, on the order of nanoliters per minute, thus allowing high MS sensitivity (7).
Most importantly, the small sample loading ability of CE allows it to be used even when the sample is limited. Even a 5 µL sample is sufficient to perform numerous injections. Unlike pressure-driven forces (such as laminar flow), CE provides a flat flow profile, and results in narrow peaks, often with a high number of theoretical plates (10). In a specific study, CE–MS performed better than LC–MS in the case of detection of hydrophilic peptides in terms of signal intensity and peak shape, and offered better separation and detection of acidic peptides, low molecular weight peptides, and shorter peptides (17). A comparison of LC–MS with CE–MS is provided in Table II.


Designing a CE–MS Method
In a previous report from our laboratory, a CE–MS based analysis of intact and digested mAbs was discussed (18). In this article, we discuss a few critical parameters that are crucial for setting up a CE–MS experiment. Sample preparation, the choice of running buffer, and optimization of MS parameters are all known to significantly influence performance, separation, and detection in CE–MS. Here, we discuss a few of these critical parameters, associated problems, their effect on resolution, and ways to improve the separation. Analysis of a typical peptide mixture is used as an example.
The position of the CE and MS instruments can have a very significant impact on achieving stable operation. The CE electrode should be at the same height as the tip of the ESI MS sprayer. Because the latter depends on the model of the MS instrument being used, the height of the CE platform must be adjusted. If this difference is greater than 1 cm, siphoning often occurs, and this leads to air inflow into the capillary, thereby resulting in abnormalities of current and improper sample injection. Also, the right side of the CE instrument must be arranged closely to the MS ion source, so as to allow a short capillary length (19,20).
Sample Preparation
Because only nanoliters of a sample are loaded when using CE, parameters such as stacking, pH junction, solid-phase extraction, cation exchange monolith, or a combination of solid phase and pH junction can all be used to achieve a high concentration in the final sample to achieve adequate signal strength (6).
Sample volume is also a critical parameter during CE–MS. Although only a nanoliter injection is required, enough sample must be present in the sample vial to ensure that the capillary touches it for pressure-assisted injections (21). Sample shortage can lead to non-reproducibility of peak areas. The sample vial must also be tightly capped to prevent loss due to evaporation, especially when working with sample volumes of less than 20 µL (22). An excess sample volume causing submergence of electrodes should also be avoided, to prevent sample carryover (21).
Correct Capillary Choice
Different types of capillaries are available for the various applications, with the bare fused silica capillary being the most widely used. However, it suffers from a significant limitation of sample adsorption on capillary walls, which could result in peak broadening and poor reproducibility. Coated capillaries that offer minimal sample wall interactions can be effective. Polyvinyl alcohol (PVA), linear polyacrylamide (LPA), and polyethylenimine (PEI) are some of the commonly used coatings for capillaries used in CE–MS (2,23). PVA-coated capillaries, for example, exhibit minimum solute wall interactions, and are stable even under basic conditions. Because of the presence of the coating, peak tailing is minimum in PVA capillaries, and reproducibility is improved (24,25). During capillary use, the sprayer end of the capillary should be cut to be smooth, with the polymer coating removed, or else sheath fluid causes it to swell and expand at the sprayer tip. Each swelling might hinder a stable nebulizing function, resulting in decreased sensitivity and fluctuations in the current. In the case of coated capillaries, burning time should be minimum, or else melting of the polymer can block the capillary (19).
Buffer Selection
The choice of running buffer, called background electrolyte (BGE), depends on the type of capillary to be used and its MS compatibility. Volatile acid-base type buffers must be used for CE–MS experiments. These include acetic acid, ammonium formate, formic acid, and ammonium acetate (19).
Use of nonvolatile buffers based on phosphoric acid or boric acid or surfactants should be avoided, because they may cause contaminants to form in the electrospray chamber, block MS ionization, and thus affect MS sensitivity. If inevitable, surfactants and additives may be used in the lowest possible concentrations, and appropriate cleaning of the MS ion source and capillary should be frequently performed (19). In a recent study, peptide samples were analyzed via CE–MS, using an acetic acid buffer and phosphate buffer, as BGE, with the same sheath fluid (13). The authors observed that, although the use of acetic acid generated good signals in the total ion electropherogram, the use of phosphate buffer produced high levels of noise, an unsteady baseline, and weakened signals. The authors suggested that it could have been due to ion pairing effects of the phosphate anions on the protonated peptides, or that they may inhibit the ionization process of peptides (13).
For analysis of therapeutic proteins, acetic acid and formic acid are commonly used as BGE, because their volatile nature makes them ESI-friendly. Also, their acidic nature facilitates ionization of intact proteins and their digested peptides, which are amenable to positive ESI (2).
The pH of the running buffer must also be carefully selected, because it dramatically affects separation in CE. High buffer concentrations are preferable because of their ability to produce a stacking effect. Stacking is a technique to preconcentrate the analyte in the capillary by applying a difference in the electric field strength between the sample and BGE. Such a difference often results in post-injection concentration of the analyte in the capillary, leading to an increase in sensitivity and production of good peak shape. Care must be taken, however, not to use an excessive concentration of buffer, because it decreases MS sensitivity (19).
The type of capillary used for separation must also be considered when deciding on the buffer pH. Coated capillaries, such as PVA, are stable in the pH range of 2.5 to 9.5 (25). Usually, 1–2% acetic acid is optimal with a PVA capillary, for peptide analysis (26,27). Like sample volume, buffer volume is also an important parameter. However, unlike the sample, which needs to be in contact with the capillary, enough buffer must be present in the buffer vial to touch the electrodes to ensure current flow (21).
Electric Field Strength
CE operates at high electric field strength, on the order of hundreds of volts (V) per cm, of the capillary. High voltage causes Joule heating if heat is not dissipated correctly, which may cause the temperature profile across the capillary cross-section to become parabolic. Such a condition leads to non-uniform flow, resulting in peak broadening and reduced resolution (28). A temperature rise may occur, which can change buffer viscosity, and affect analyte separation. One degree change in temperature may cause more than a 2.5% change in analyte mobility (10). It is thus essential to maintain temperature during CE–MS operations. Effective heat dissipation also depends on the capillary diameter. A capillary with a narrow inner diameter and wide outer diameter offers better heat dissipation. CE resolution is also directly dependent on the applied voltage. However, it should be noted that resolution is proportional to the square root of the applied voltage; therefore, doubling the voltage does not double the resolution (10).Figure 2 illustrates the separation profile of a peptide mixture at electric field strengths of 300 V/cm and 600 V/cm. The improved signal intensity, additional peaks, and a better resolution at 600 V/cm are likely due to the direct relationship between the resolution and the applied voltage. Higher voltages also result in shifts of migration times towards the origin, reducing the net analysis time. An improvement in the separation profile also suggests the effective dissipation of heat from the capillary, due to a suitable temperature control system in the instrument, noting that cassette temperature was set at 20 °C during the analysis.

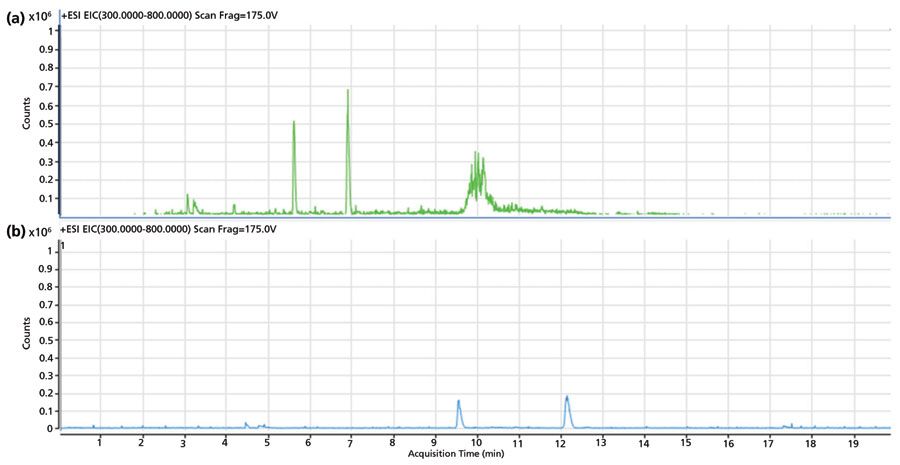
Figure 2: Effect of applied electric field strength on CE–MS separation profile. Extracted ion chromatograms (EIC) of sample A performed at (a) 600 V/cm, and (b) 300 V/cm on a PVA capillary using 2% acetic acid as BGE.
Sheath Fluid
Typically, a freshly prepared 50% methanolic solution alone, or 5 mM ammonium acetate in a 50% methanolic solution, yields satisfactory results, with both positive and negative polarity modes of CE–MS. However, before use, it is crucial to degas the sheath fluid, or else air bubbles may lead to the appearance of spike-like signals on the electropherogram. Another caution is to maintain the ambient temperature, which may affect the viscosity of the sheath liquid. A change in temperature may cause a change in the viscosity of the sheath liquid solution, thereby causing the pressure of the sheath liquid sending pump to oscillate and change the flow rate (19).
Sample Injection
The amount of sample injected also affects the separation profile in CE. Although loading too little of the sample may compromise sensitivity, care must be taken to avoid sample overloading. In CE sample loading, injection plug length is more critical than injection volume. An excess injection can cause peak broadening, which decreases separation efficiency. Also, the difference in conductivities of the sample and the buffer can produce field inhomogeneities and distorted peak shapes. The typical practice is that the plug length should be 1–2% of the capillary length. The volume of sample loaded is a function of capillary dimensions, buffer viscosity, applied pressure, and time. When using hydrodynamic injection, normal pressure and time should range from 25 to 100 mbar and 0.5 to 5 s, respectively (6,10).
Improving Resolution
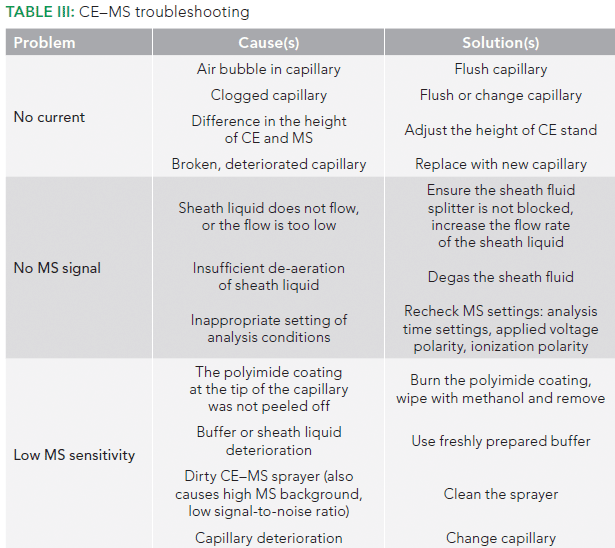
In case multiple overlapping peaks are obtained in a given period, the separation may be enhanced by decreasing the electric field, which slows down the separation, thereby providing an opportunity for generation of the tandem spectra. The capillary length may also be increased to slow down separation and improve resolution. In the case of peptides, the separation pH may also be raised. High pH will cause partial deprotonation of aspartic and glutamic acid residues in addition to basic groups in peptides. The acquisition of a partial negative charge on peptides will lead to slower and wider separations (6). Some other problems that might be encountered during CE–MS analysis, along with their causes and solutions, are listed in Table III.

Conclusions
Various parameters must be considered while designing a CE–MS experiment, in order to harness the potential of the technique fully. These considerations are of utmost importance, and may directly or indirectly affect the performance of the system. In the future, the introduction of protein or peptide-specific migration standards, and the development of in silico methods for predicting CE migration behavior under different analysis conditions, might significantly enhance the usage of CE–MS. Such developments would all be to the benefit of the scientific community in the field of biotherapeutics. Development of microchip-based CE–MS (29) is another exciting development to look for in the future.
References
(1) J.R. Lill, Introduction to Biotherapeutics in Analytical Characterization of Biotherapeutics (John Wiley & Sons, Hoboken, New Jersey, 1st ed., 2017), pp. 1–14.
(2) S.S. Zhao and D.D.Y. Chen, Electrophoresis 35(1), 96–108 (2014).
(3) G. Walsh, Nat. Biotechnol. 36(12), 1136–1145 (2018).
(4) A.S. Rathore, Trends Biotechnol. 27(12), 698–705 (2009).
(5) A.S. Rathore, I.S. Krull, and S. Joshi, LCGC North Am. 36(11), 814–822 (2018).
(6) L. Sun, G. Zhu, X. Yan, Z. Zhang, R. Wojcik, M.M. Champion, and N.J. Dovichi, Proteomics 16(2), 188–196 (2016).
(7) K. Faserl, B. Sarg, P. Gruber, and H.H. Lindner, Electrophoresis 39(9–10), 1208–1215 (2018).
(8) M. Pioch, S.C. Bunz, and C. Neusü , Electrophoresis 33(11), 1517–1530 (2012).
(9) R.L.C. Voeten, I.K. Ventouri, R. Haselberg, and G.W. Somsen, Anal. Chem. 90(3), 1464–1481 (2018).
(10) D. Heiger, High Performance Capillary Electrophoresis, Agilent Technologies (2000).
(11) G. Rozing, Rozing.com Consulting at <https://www.rozing.com/>
(12) J.M. Risley, C.A.G. De Jong, and D.D.Y. Chen, Electrospray Ionization Interface Development for Capillary Electrophoresis – Mass Spectrometry in Capillary Electrophoresis–Mass Spectrometry (CE-MS): Principles and Applications (John Wiley & Sons, Hoboken, New Jersey, 2016), pp. 7–39.
(13) M. Serwe and G.A. Ross, A Comparison of CE–MS and LC–MS for Peptide Samples, Agilent Technologies, 2000.
(14) H. Mischak, J.N. Joshua J. Coon, E.M. Weissinger, J.P. Schanstra, and A.F. Dominiczak, Mass Spectrom. Rev. 28(5), 703–724 (2009).
(15) E. Dominguez-Vega, T. De Vijlder, E.P. Romijn, and G.W. Somsen, Anal. Chimi. Acta 982, 122–130 (2017).
(16) G. Zhu, L. Sun, J. Heidbrink-Thompson, S. Kuntumalla, H. Lin, C.J. Larkin, J.B. McGivney IV, and N.J. Dovichi, Electrophoresis 37(4), 616–622 (2016).
(17) C.S. Babu, CE/MS and LC/MS Synergy Complementary Solutions for Peptide Mapping (Agilent Technologies, 2013).
(18) S.K. Singh, V. Hebbi, A.S. Rathore, and I.S. Krull, LCGC North Am. 34(9), 720–728 (2016).
(19) Agilent Technologies, CE/MS Analysis Optimization Guide (2009).
(20) Installing the Agilent CE ESI–MS Sprayer Kit (Agilent Technologies, 2000).
(21) Agilent Technologies, Agilent 7100 Capillary Electrophoresis System (2017).
(22) Agilent Technologies, High Performance Capillary Electrophoresis: A Primer (Agilent, 2nd Ed., 2014).
(23) J. Q. Xia, Coated Capillaries for CE-MS of Therapeutic Protein in Capillary Electrophoresis-Mass Spectrometry: Therapeutic Protein Characterization (Springer International Publishing, Switzerland, 2016), pp. 7–11.
(24) D. Belder, A. Deege, H. Husmann, F. Kohler, and M. Ludwig, Electrophoresis 22(17), 3813–3818 (2001).
(25) Agilent Technologies, Polyvinyl Alcohol (PVA) Coated Capillaries at <https://www.agilent.com/en/products/capillary-electrophoresis-ce-ms/ce-ce-ms-supplies/capillaries/polyvinyl-alcohol-(pva)-coated-capillaries>
(26) Agilent Technologies, Identification and Quantification of Oxidation Sites on Monoclonal Antibodies Using Capillary Electrophoresis and Quadrupole Time-of-Flight Mass Spectrometry (2013).
(27) Agilent Technologies, Glycopeptide Analysis of Antibodies by Capillary Electrophoresis and Q-TOF (2011).
(28) A.S. Rathore, J. Chromatogr. A 1037(1–2), 431–443 (2004).
(29) G. Rozing, Recent Developments of Microchip Capillary Electrophoresis Coupled with Mass Spectrometry in Capillary Electrophoresis-Mass Spectrometry (CE-MS): Principles and Applications (John Wiley & Sons, Hoboken, New Jersey, 2016), pp. 67–102.
ABOUT THE COLUMN EDITOR

Ira S. Krull

Ira S. Krull is a Professor Emeritus with the Department of Chemistry and Chemical Biology at Northeastern University in Boston, Massachusetts, and a member of LCGC's editorial advisory board.
ABOUT THE AUTHOR

Ramesh Kumar

Ramesh Kumar is a graduate student at the Department of Chemical Engineering at the Indian Institute of Technology in Delhi, India.

Anurag S. Rathore

Anurag S. Rathore is a professor in the Department of Chemical Engineering at the Indian Institute of Technology in Delhi, India.




















, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.




