Data Integrity Focus, Part VI: Who Is Looking Over Your Shoulder? Quality Oversight for Data Integrity
LCGC North America
What are the roles and responsibilities of quality assurance staff for ensuring data integrity in an organization?
The focus of this series so far has been on the chromatography laboratory. In this last part, we look at the roles and responsibilities of quality assurance staff for ensuring data integrity in an organization.
This is the sixth article in the data integrity focus series. The first article presented and discussed a data integrity model to present the scope of data integrity and a data governance program for an organization (1). The second part discussed data process mapping to identify data integrity gaps involving a chromatography data system (CDS), and looked at ways to remediate them (2). The operation of a CDS as a hybrid system was the subject of the third article (3), and the fourth discussed how complete data and raw data mean the same thing for records created in a chromatography laboratory (4). The fifth part looked at how the updated USP<1058> general chapter on analytical instrument qualification (AIQ) can help with ensuring data integrity with a CDS (5). Here, our attention will turn to the quality assurance (QA) oversight necessary to ensure data integrity.
Quality Does Not Own Quality Anymore
Before we start to look at the role of quality assurance in data integrity, it is important to remember that Quality Control (QC) or QA does not own quality anymore. The reason? Quality is everybody's responsibility. This is the foundation of the data integrity model, presented and discussed in Part 1 of this series (1). Given that data integrity is an integral part of data quality, it is the responsibility of the testers, reviewers, and management in a regulated laboratory to ensure that data are complete, consistent, and accurate, and that the principles of the ALCOA+ concept–attributable, legible, contemporaneous, original, and accurate; plus, complete, consistent, enduring, and available–have been applied to the data. The European Union good manufacturing practices (EU GMP) regulations for laboratory records require that the performer and reviewer of analytical data ensure that the work has taken place (6), that data are complete, accurate, and comply with applicable procedures and regulations (7,8), and that checks have been made to determine if data have been falsified (9–11). These steps are the responsibility of laboratory staff. Knowing that you have made errors, but having an attitude that "QA will sort it out" is neither professional nor compliant with the regulations.
Quality Control Unit or Quality Assurance?
One of the problems with discussing the role of a quality function is the wording of regulations. The U.S. Food and Drug Administration (FDA) GMP regulations in Section 211.22 require a quality control unit (7). The issue is that in many FDA warning letters, the quality control unit has mutated into the quality unit; the FDA regulations are showing their age. In contrast, EU GMP specifies quality assurance in Chapter 2 on personnel (12), and one of the responsibilities is self-inspections under EU GMP chapter 9 (13). In this column, when the term QA is used, it means an independent quality assurance unit or the quality unit, and not a laboratory function. So, what does the QA function do in an organization?
Quality Oversight
What does quality oversight mean in practice? Because English is such a wonderful language, there are two definitions for oversight:
- responsibility for making sure an activity is being done correctly
- a mistake made because of a failure to notice something.
Which one do you think applies to the QA department? Obviously, the first definition is applicable, and this is QA's primary role. There must be checks to ensure that work, including following procedures, collecting and reporting data, and documenting have been performed correctly. Could QA be involved with the second definition? Yes, unless your laboratory never has any deviations or out-of-specification (OOS) results. Therefore, there will be quality oversight checking that laboratory deviations and OOS results have been correctly investigated, the root causes identified, and that corrective and preventative action (CAPA) plans are implemented. QA will also be involved in monitoring the effectiveness of preventative action plans, to see that the same deviation does not occur again.
Understanding the Cost of Non-Compliance
Why should QA be involved in oversight activities? Quality assurance is a function that must be independent of the laboratory. However, in many warning letters and Form 483 observations, it is the failure of QA to identify and remedy falsification or poor data management practices that is a root cause of many instances of non-compliance. To understand this better, we need to consider the cost of compliance (cost of doing it right) versus the cost of non-compliance (the cost of getting caught), as shown in Figure 1.


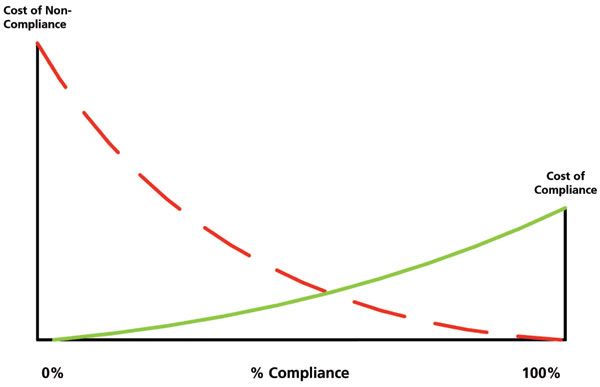
Figure 1: Balancing the cost of compliance and non-compliance (14). Reproduced with permission.
The horizontal axis is the percentage of compliance from 0 to 100%. The only fixed points are at the ends of the scale where 0% is where the company is out of compliance, and no controls exist, to the other extreme of 100%, where anything that can be compliant is compliant. In the latter situation, not much work may be done, but it is fully compliant.
The left-hand vertical axis is the cost of non-compliance, and the right-hand axis is the cost of compliance. You will note the cost of compliance axis is smaller than the cost of non-compliance axis. This is one of the balances you need to consider. Fixing a regulatory problem that has been identified in an inspection is always more expensive than doing the right job, or finding a problem and fixing it yourself. I suggest you read a consent decree such as that for Ranbaxy (15), or look at some data integrity warning letters that have resulted in import alerts. The cost of non-compliance can now be quantified as hundreds of millions of dollars for some companies.
Effective risk management is essential to determine where your laboratory operations are in Figure 1. QA is there to see that risk mitigation is effective and is compliant with both laboratory procedures and the company's interpretation of the regulations.
Role of QA in a Data Integrity Program
From Part 1 of this series (1), the data integrity model shown on included quality assurance activities. There, it was noted that QA is involved in:
- compliance oversight
- data integrity audits
- data integrity investigations.
Of these three, most chromatographers are aware of compliance oversight: checks by QA over and above the four-eyes review carried out by the laboratory. However, fewer readers will be aware of what data integrity audits and investigations involve, and those are the focus of this column. One potential problem comes with the great and the good of all regulated organizations: senior management. The additional requirements for data integrity audits and investigations place an increased burden on QA resources. If management does not ensure that there are adequate resources available, we can fall back to a discussion about the cost of non-compliance when an inspector finds a data integrity violation.
It is interesting to note that, in many companies where a lean approach to compliance is taken, there is never any money available for compliance work, or it must be phased in over a long period. However, if there is an adverse regulatory finding, money suddenly flows like Niagara Falls.
The aim of any regulated laboratory should be to remain in compliance with effective risk management. Please note that risk management is not an excuse to do nothing, but to put scarce resources where they are most needed.
Prevention is Better than a Cure
To ensure that a company remains in compliance, there is a regulatory expectation that QA staff perform data integrity audits as part of their oversight activities. This expectation was set by the Medicines and Healthcare products Regulatory Agency (MHRA) web site in December 2013 (16), and is a feature of several data integrity guidance documents published since then. Part of the oversight activities of QA are audits to ensure that data generated for good practice (GXP) purposes are complete, consistent, and accurate, and complies with the ALCOA+ principles.
The expectations for both paper and electronic records are best explained in Appendix 1 of the WHO guidance (9), where they are set against ALCOA principles. The latest draft of the Pharmaceutical Inspection Co-operation Scheme (PIC/S) guidance has sections on the regulatory requirements for both paper and electronic records (11). For hybrid systems, which were discussed in Part 3 (3), you'll need to merge the requirements for both record formats, and you can see why you should work electronically (17). These are the criteria that can be used for data integrity audits of paper processes and computerized systems. It is important to remember that there are many manual processes in laboratories, such as sample preparation, where many data integrity vulnerabilities can be identified (18).
Planning and Scheduling Audits
Consistent with any auditing program, data integrity audits need to be planned and scheduled. Some of the inputs into this process are:
- a current inventory of computerized systems and processes that is classified by risk to data and patient
- assessment of these processes, and systems to identify and remediate data vulnerabilities
- identification of key processes and systems where procedural controls are used for remediation
- identification of critical validated computerized systems (even if technical controls are used for electronic working), such as CDS, laboratory information management systems (LIMS), and the electronic laboratory notebook (ELN).
The aim is to identify those processes and systems with records and data that are the highest risk and regulatory scrutiny. From these and other inputs, process and systems are rated according to criticality and importance, and a schedule drawn up for their audit. One major pharmaceutical company conducts a data integrity audit of their global CDS every three months, due to importance of the data and regulatory scrutiny. Each QA group makes its own plans and schedules based on risk, data vulnerability, and resources, but remember when doing this to keep in mind Figure 1.
The Data Integrity Model in Audit Preparation
When preparing for a data integrity audit, consider how the data integrity model can help, and what assumptions are being made (and possibly challenged) in the audit preparation:
- Foundation: Management leadership, open culture, data integrity policy, and good documentation practices for paper, hybrid, and electronic systems, plus records of staff training and understanding
- Level 1: Qualification of analytical instruments, and validation of computerized systems and software, including spreadsheet applications. Instruments and systems are fit for their intended use
- Level 2: Development of robust analytical procedures that are verified under actual conditions of use
- Level 3: Application of all lower levels to the analysis of samples, and generation of data that are complete, consistent, and accurate
The focus of a data integrity audit in a regulated laboratory is at Level 3, but be prepared to request records and data from the lower levels to ensure that analytical work has been carried out correctly.
Scope of a Data Integrity Audit
It is important to realize that a laboratory data integrity audit is not simply having a look at a computerized system. There are many places in the analytical process where falsification can be hidden, and that does not include a computerized system, as shown in Figure 2. Sampling and sample preparation are two main areas where changes can be made without any record, because these are manual processes, and therefore it is very difficult to identify any repeat work (18).

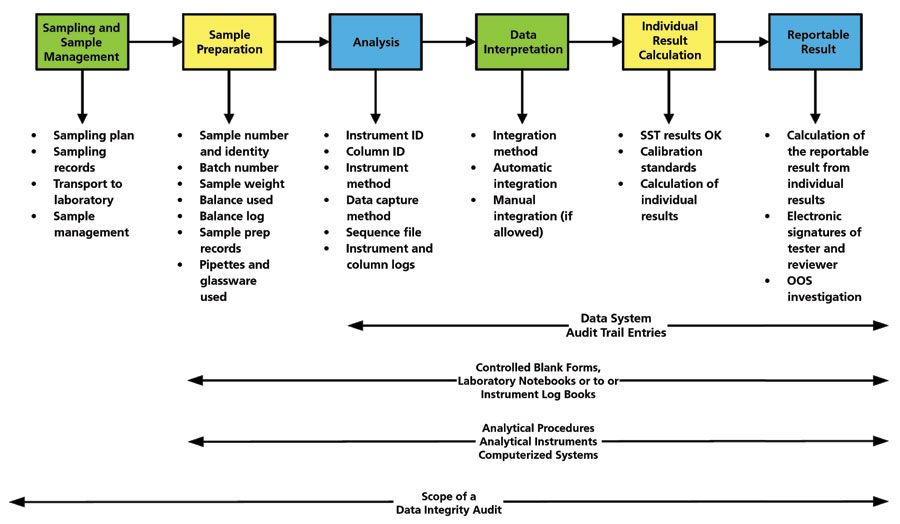
Figure 2: Scope of a data integrity audit in a chromatography laboratory (17). Reproduced with permission.
The basic premise of a data integrity audit is that the auditor should be able to trace from the sample to a reportable result easily and transparently if all records are available, complete, and accurate. The same applies in reverse; an auditor should be able to pick a reportable result and be able to trace backwards to the sample.
As the FDA states in its 1993 Guide to Inspection of Pharmaceutical Quality Control Laboratories (19):
Laboratory records and logs represent a vital source of information that allows a complete overview of the technical ability of the staff and of overall quality control procedures.
As the audit progresses and an instrument, computerized system, or analytical method is found, the auditor can delve in the qualification, computer validation, or calibration checks to find out further information. If required, training records of analytical staff can be requested. When the applicable procedures and specifications are identified, they can be read to see how they have been followed by the analyst and the second-person reviewer, if the individual results and reportable result comply, respectively.
Remember that audits sample; the focus is on checking that procedures work correctly, as defined in the first oversight definition seen earlier:
- Are all records and data complete, consistent, and accurate?
- Are data transformations transparent?
- Where data have been entered manually into a computerized system, are the input figures correct, or if corrected, does the audit trail contain the old value, and the reason for change?
- Where peaks have been manually integrated, has this been done correctly?
- Are all analysts identified?
- Are all handwritten records legible, and follow good documentation practices?
- Have the audit trail entries been reviewed? If review by exception has been used, look at the entries to confirm that no changes have been made or data have been deleted.
Where a computerized system is involved, the data integrity audit also needs to spot check items, such as appropriate access privileges for both users and administrators, and that the application configuration settings still enforce data integrity. In addition, all data integrity guidance documents require checks for any unofficial testing, falsification, or copying of data (9–11,20), both inside the application but also in the recycle bin.
Audit Checklist or Aide-Memoire?
How should a data integrity audit, or indeed any audit, be conducted? Many will say to use a checklist. The problem is that a checklist does not work; it leads the auditor as he or she tries to complete the whole document. Pity the poor auditee at question 257 who is comatose with boredom. Instead, an aide-memoire, or memory aid, should be used. Ah, you may think, I'm just playing with words, it's really a camouflaged checklist.
A checklist is a fixed set of questions, in comparison to an aide-memoire that has more general and broad questions and allows an experienced auditor to have a more natural conversation with the auditees, deviate where necessary, and be under no pressure to complete a list of questions.
Are Data Integrity Audits and Periodic Reviews the Same?
Chromatography data systems are a prime target in regulatory inspections, and should rightly be the subject of regular data integrity audits. However, computerized systems also require periodic reviews (21), raising the question of whether there is any overlap between the two types of audits. Let us look at the focus of each one:
- In a periodic review, computerized systems should be periodically evaluated to confirm that they remain in a valid state, and are compliant with GMP. Such evaluations should include, where appropriate, the current range of functionality, deviation records, incidents, problems, upgrade history, performance, reliability, security, and validation status reports (21).
- In a data integrity audit, routine audits or self-inspections of computerized systems may reveal gaps in security controls that inadvertently allow personnel to access and potentially alter time and date stamps (9). There are other quotations on the topic available!
As you can see, the aims of the two are different. A periodic review asks: Does the system remain validated? An audit asks: Are there any data integrity gaps or violations? However, there are overlaps, as can be seen in Table I. The question is to ensure that these areas do not fall between the gaps, or get repeatedly examined each time an audit or review takes place.

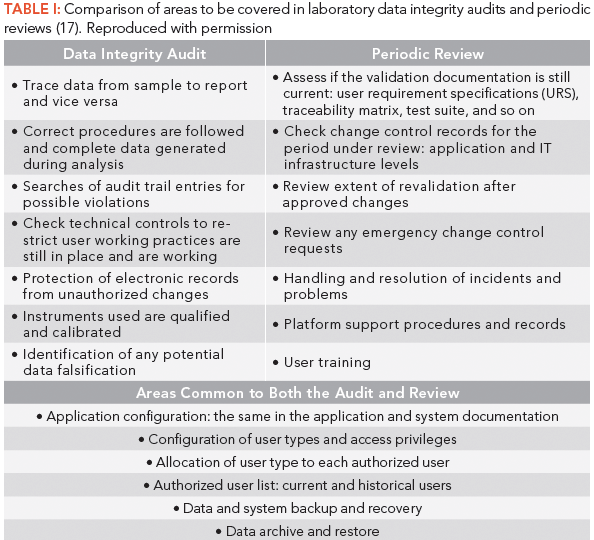
Table I: Comparison of areas to be covered in laboratory data integrity audits and periodic reviews (17). Reproduced with permission
The aim of a data integrity audit is to identify any poor data management practices and data vulnerabilities, as well as any evidence of data falsification. In the first two cases, these will lead to corrective and preventative action plans to remediate and provide a long-term solution to prevent reoccurrence. In the case of data falsification, this must trigger a data integrity investigation.
What is a Data Integrity Investigation?
If a data integrity audit, regulatory inspection, or a whistleblower identifies a data integrity violation, the next step is a data integrity investigation (9–11). It is important to understand that data integrity investigations are not always caused by scheming scientists huddled in smoke filled rooms (where still permitted by local health and safety laws), plotting how to falsify data, but an integrity issue can also be caused by software bugs (17). We will focus on data integrity investigations involving falsification. Question 18 in the FDA data integrity guidance document is the best place to start, because it discusses hiring a third-party expert for independence, and links to the FDA's 1991 Application Integrity Policy (22). The scope of a data integrity investigation is shown in Figure 3. Because this is very extensive, we will only present the main points, as there is not sufficient space to discuss the whole spectrum of work involved here. For more information, please see a recent publication on the subject (17).

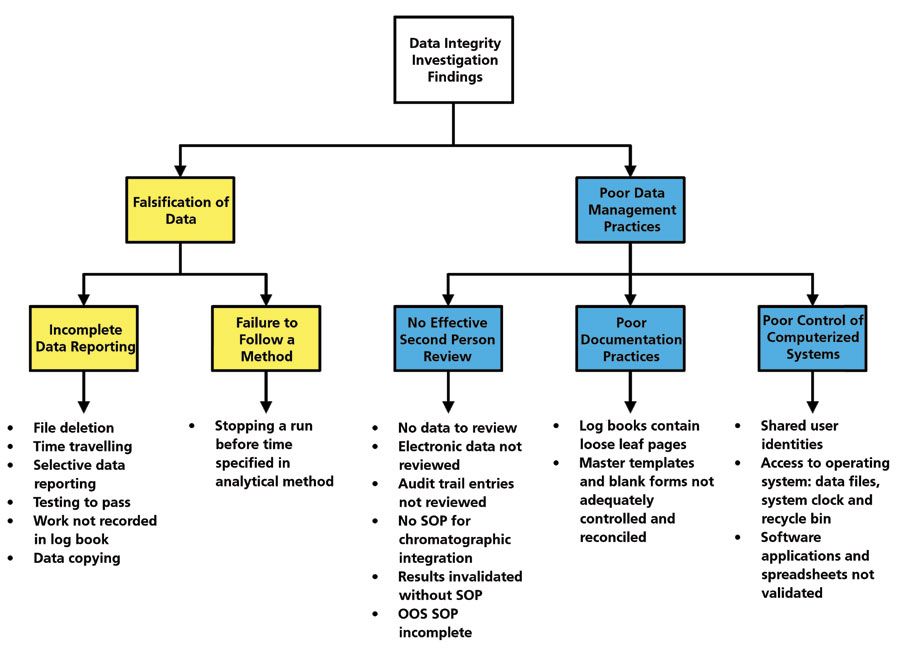
Figure 3: Scope of a data integrity investigation (17). Reproduced with permission.
Some of the activities that need to be performed are:
- Sequester a copy of the impacted data, data sets, or database, or take the system out of service for the duration of the investigation.
- Interview staff, including current and former employees, to understand why the data integrity violations occurred.
- Determine the extent of the problem.
- Conduct a detailed investigation of processes and systems to identify the violations.
- Define and justify the time frame of the data integrity violations.
- Identify the processes, systems, and products affected by the data integrity violations, such as production, quality control, or quality assurance.
- Make a comprehensive assessment of the impact of the violations, including a risk assessment on the products, the patient, or regulatory submission(s).
Data Integrity Investigation Case Study
During the preparation for an FDA pre-approval inspection (PAI), the data generated from three batches of product submitted in a regulatory submission application were reviewed to ensure that the data were complete, consistent, and accurate. During this review, some discrepancies were found between entries in the instrument log books and chromatography data system records (both electronic records and paper printouts) used to release the three batches. The initial investigation revealed the following problems and inconsistencies with data and records:
- In some chromatographic analyses, more system suitability test (SST) injections had been made than defined by USP <621> (23) and the laboratory standard operating procedure (SOPs).
- Some injections were not documented in the instrument log book, but exist in an instrument data system.
- Some injections in the data systems have duplicate times, indicating that they were injected at the same time, but the log book only shows a single set of injections.
- Data have been copied from earlier analysis and used for later work in validating an analytical method.
The discrepancies appeared to be due to a single individual who left the company when these issues were raised. Thus, a data integrity investigation was started to determine the extent and impact of these data integrity breaches. One of the requirements of a data integrity investigation is the determination of material impact.
Any organization undertaking a data integrity investigation will expend a large amount of time and resources. It is also important that the maximum benefit be obtained from this investment in resources. You may disagree about the use of the word "benefit" in the last sentence, but look at the investigation as an opportunity to clean up not just the violation, but also identify other poor data management practices that may exist in a laboratory. This is to ensure that not only the current issue is remediated and resolved, but that poor practices are eliminated that could cause violations in the future. Due diligence is required to find not just the root causes of the violation, but also to identify and include in the CAPA plan those poor data management practices that could contribute both directly and indirectly to the violations now and in the future.
Any data integrity investigation is, of necessity, detailed and slow, because there are many items to check and cross check to identify any potential violation. The primary areas to check are generally:
- paper records for sampling and sample preparation
- records in the chromatography data system versus printouts versus instrument log books
- manual data entries into computerized systems
- records of the second person review
- recycle bins on computerized systems and can users access the system clock
Secondary areas to check include:
- company approach to data integrity, and procedures for ensuring data integrity with training records and open culture
- qualification and calibration of analytical instruments
- validation of computerized systems, including configuration settings; are technical controls used effectively?
- analytical procedure development and validation records.
As you can see, the investigation can cover all levels within the data integrity model that we discussed in the first part of this series (10). As shown in Figures 3 and 4, the work performed needs to be documented in a data integrity investigation report listing the scope of the work performed, the findings of falsification and poor data management practices, root causes of the violations, and the CAPA plans for corrective and preventative measures. A summary of the investigation findings is shown in Figure 4, and you'll note that there were more poor data management practices identified than instances of data falsification. One important section that must be present in the report is a statement of material impact.

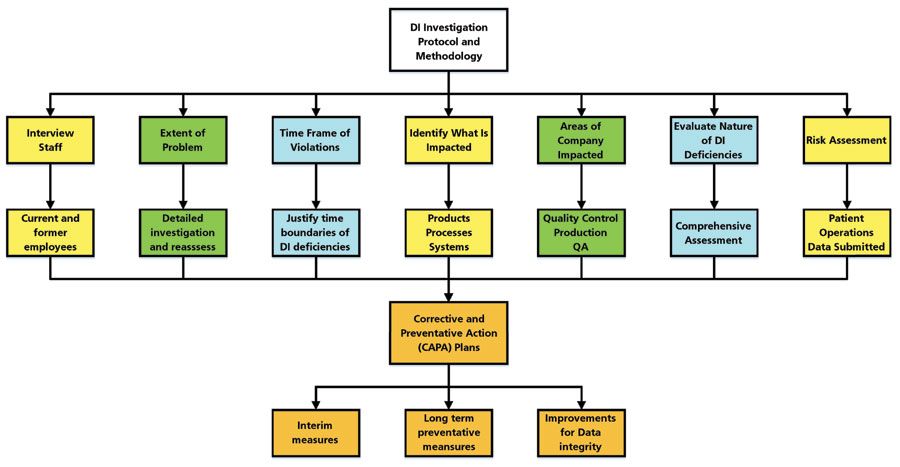
Figure 4: Summary of a data integrity investigation showing data falsification and poor data management practices (17). Reproduced with permission.
Assessment of Material Impact
One of the outcomes of a data integrity investigation is to determine if there is any material impact of the falsification on a regulatory submission, product quality, or patient safety. Material impact is a legal term meaning that there is an adverse impact on a regulatory status or product quality (such as, for example, releasing a drug product that is under or over strength, or a regulatory dossier containing falsified data). Material means a significant factor in a decision, as opposed to a trivial or unimportant one. It is essential that this determination be made at the end of an investigation to decide if regulatory authorities need to be informed. The rationale for this decision must be included in the investigation report. The PIC/S guidance document notes that it is better to disclose to a regulator these issues, rather than just sit on the report (11).
Summary
Quality oversight is an important component of an overall data integrity program within any regulated organization, to ensure that work has been performed correctly. Where problems have been identified, there need to be preventative, and corrective plans put in place. In the case of a data integrity violation, quality assurance needs to conduct a thorough and detailed investigation that will determine the extent and impact of any falsification, and determine if there is any material impact on a regulatory submission, product quality, or patient safety.
References
(1) R.D. McDowall, LCGC North Amer. 37(1), 44–51 (2019).
(2) R.D. McDowall, LCGC North Amer. 37(2), 118–123 (2019).
(3) R.D. McDowall, LCGC North Amer. 37(3), 180–184.
(4) R.D. McDowall, LCGC North Amer. 37(4), 265–268.
(5) R.D. McDowall, LCGC North Amer. 37(5), 312–316.
(6) EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 1 Pharmaceutical Quality System (European Commission, Brussels, Belgium, 2013).
(7) 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products (Food and Drug Administration: Silver Springs, MD, 2008).
(8) EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 6 Quality Control. (European Commission, Brussels, Belgium, 2014).
(9) WHO Technical Report Series No.996 Annex 5 Guidance on Good Data and Records Management Practices. (World Health Organization, Geneva, Switzerland, 2016).
(10) FDA Guidance for Industry Data Integrity and Compliance With Drug CGMP Questions and Answers (Food and Drug Administration, Rockville, MD, 2018).
(11) Pharmaceutical Inspection Co-operation Scheme/Pharmaceutical Inspection Convention, Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments (Geneva, Switzerland, Draft, 2018).
(12) EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 2 Personnel.(European Commission, Brussels, Belgium, 2014).
(13) EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 9 Self Inspection (European Commission, Brussels, Belgium, 2001).
(14) R.D. McDowall, Validation of Chromatography Data Systems: Ensuring Data Integrity, Meeting Business and Regulatory Requirements (Royal Society of Chemistry, Cambridge, United Kingdom, 2nd Ed., 2017).
(15) Ranbaxy Laboratories Ltd & Ranbaxy Inc: Consent Decree of Permanent Injunction. 2012.
(16) MHRA Expectation Regarding Self Inspection and Data Integrity 2013 [cited 2013 01 Jan 2016]; Available from: http://webarchive.nationalarchives.gov.uk/20141205150130/http://www.mhra.gov.uk/Howweregulate/Medicines/Inspectionandstandards/GoodManufacturingPractice/News/CON355490.
(17) R.D. McDowall, Data Integrity and Data Governance: Practical Implementation in Regulated Laboratories. (Royal Society of Chemistry, Cambridge, United Kingdom, 2019).
(18) M.E. Newton and R.D. McDowall, LCGC North Amer. 36(1), 46–51 (2018).
(19) Barr Laboratories: Court decision strengthens FDA's regulatory power. 1993; Available from: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/Manufacturing/ucm212214.htm.
(20) MHRA GXP Data Integrity Guidance and Definitions (Medicines and Healthcare Products Regulatory Agency, London, United Kingdom, 2018).
(21) EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 11 Computerized Systems (European Commission, Brussels, Belgium 2011).
(22) US Food and Drug Administration, FDA Application Integrity Policy: Fraud, Untrue Statements of Material Facts, Bribery, and Illegal Gratuities Compliance Policy Guide Section 120.100 (Food and Drug Administration, Rockville, MD, 1991).
(23) USP General Chapter <621> Chromatography. (United States Pharmacopoeia Commission Inc., Rockville, MD, 1991).
R.D. McDowall is the director of R.D. McDowall Limited in the UK. Direct correspondence to: rdmcdowall@btconnect.com



















Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.


Analyzing Vitamin K1 Levels in Vegetables Eaten by Warfarin Patients Using HPLC UV–vis
April 9th 2025Research conducted by the Universitas Padjadjaran (Sumedang, Indonesia) focused on the measurement of vitamin K1 in various vegetables (specifically lettuce, cabbage, napa cabbage, and spinach) that were ingested by patients using warfarin. High performance liquid chromatography (HPLC) equipped with an ultraviolet detector set at 245 nm was used as the analytical technique.

Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.


