HILIC and Its Applications for Biotechnology, Part II
LCGC North America


The modern usage and specific applications of HILIC for two other major classes of analytes - glycopeptides and glycoproteins - are discussed.
Here we describe novel application areas in which hydrophilic interaction liquid chromatography (HILIC) has vast importance and utility, including peptides, glycopeptides, two-dimensional liquid chromatography in proteomics, oligonucleotides, lipids and lipidomics, glycan analysis, proteins, glycoproteins, phosphoproteins, and related biopolymers. We also describe some specific HILIC conditions that have proven useful for these classes of biologically active analytes and provide typical chromatograms from the recent literature, both scientific and commercial.
In part I of this two-part series on modern hydrophilic interaction liquid chromatography (HILIC) applications of relevance to the biotechnology industry, we discussed the fundamental principles and mechanisms suggested from the literature for this, now very widely accepted and practiced form of modern high performance liquid chromatography (HPLC) and ultrahigh-pressure liquid chromatography (UHPLC). We also discussed just a few of the common applications already described for simple and complex glycans often derived from glycopeptides or glycoproteins or for synthetic, standard glycans. We discussed the current roles of HILIC in glycoprofiling and glycan analysis from any source, but in particular by deglycosylation of proteins and peptides. In part II, we discuss modern usage and specific applications of HILIC for two other major classes of analytes important in biotechnology today: glycopeptides and peptides, and glycoproteins and proteins. Other types of proteins, such as phosphoproteins and their applications, are also discussed. Finally, we propose some possible future applications in which HILIC can prove itself useful, including complex glycans, glycomics, oligo-drugs (for example, heparins), glycosaminoglycans, and others.
Peptides
HILIC stationary phases were developed with the aim of providing a tool for the analysis of hydrophilic compounds, which are poorly retained in reversed-phase liquid chromatography (LC) and, therefore, are not ideally resolved (1). Although the elution order of analytes in HILIC is generally reversed compared to that in reversed-phase LC (that is, the most hydrophobic compounds are eluted first in HILIC and the most hydrophilic are eluted last), it has been recognized that the retention order is not inversely proportional (2). Because of various interactions in the HILIC mode that contribute to the separation and selectivity in reversed-phase LC and HILIC, the techniques to a large degree are orthogonal (3,4).
Besides the analysis of small molecules, many early HILIC applications investigated the separation of peptides. Some of these include those of Alpert (1), Yoshida (5), and Mant and colleagues (6). More recently, Gilar and Jaworski (3) compared three HILIC stationary phases and concluded that for the separation of charged species, such as peptides, the interactions with the charged silanols on silica significantly contributed to the retention mechanism.
The separation orthogonality of HILIC is often used for the analysis of compounds that are difficult to resolve in reversed-phase LC. For example, Zhu and colleagues (2) have used both reversed-phase LC and HILIC for analyses of peptide maps of erythropoietin and found that the methods were complementary. HILIC was more suitable for the detection and quantification of small or hydrophilic peptides and also glycosylated peptides that were not adequately resolved in reversed-phase LC. Moreover, HILIC peptide maps contained some unique, very hydrophobic peptides that were not detected in reversed-phase LC.
HILIC was also applied to the quantitative bioanalysis of therapeutic peptides and proteins. This class of biological therapeutics holds great potential; however, the analysis has been challenging. Biological fluids contain many peptides, especially when the analysts perform proteolysis of serum proteins before liquid chromatography–mass spectrometry (LC–MS). Two-dimensional liquid chromatography (2D LC), reversed phase × HILIC, combined with multiple reaction monitoring mass spectrometry (MS), was used for the quantitation of peptides of interest. Limits of detection below 10 ng/mL were achieved (7).
As stated above, HILIC and reversed-phase LC separation modes are orthogonal, and they can be utilized for 2D LC. D'Attoma and colleagues (8) adopted HILIC as the second dimension of 2D LC for the separation of protein digests. They reported an effective peak capacity of 2600 in a 180-min analysis. A HILIC × reversed-phase 2D LC setup was also evaluated by Di Palma and colleagues (10) for the separation of complex proteomic samples. The authors observed a favorable separation of peptides on zwitterionic HILIC in the first dimension compared to the more typical, first dimension — strong cation-exchange chromatography. Importantly, the authors noted a potential for enrichment of phosphopeptides in HILIC × reversed-phase 2D LC (9,10). McNulty and Annan (11) also investigated the potential of HILIC × reversed-phase 2D LC for proteomic analysis (11). Similar to previous reports, they observed significant advantages of HILIC for phosphoproteomic analysis. Because of charge–charge interactions with the zwitterionic stationary phase, or simply because of the more hydrophilic nature caused by phosphate groups, the phosphopeptides were eluted in a later elution time window when compared to general peptides (9–11). Phosphopeptides also appeared to be better recovered and more detectable in the HILIC × reversed-phase 2D LC method (11). A similar observation was made by Alpert (12), who proposed that the solute's interaction with the charged, hydrophilic sorbent may affect their elution patterns. This separation mode was then termed electrostatic repulsion hydrophilic interaction chromatography (ERLIC), and it can be used for the selective isolation of phosphopeptides from a protein digest. To further this observation, Sze (13,14) has used ERLIC–weak anion-exchange approaches (HILIC on weak anion-exchange surfaces) for selectively retaining phosphopeptides, free of other, acidic peptides, by operating at pH 2, where the phosphate group retains a negative charge and peptide carboxyls are neutral.
Oligonucleotides
HILIC was recently applied to the separation of another class of therapeutic molecules: oligonucleotides. These molecules (with a hydrophilic, phosphate backbone) were separated on a prototype hydroxymethyl methacrylate based monolithic column (15), or on commercially available HILIC columns (16). The mobile phase included triethylammonium acetate (TEAA), an ion-pairing system often used in reversed-phase LC. However, it is not clear to what extent the ion pairing contributes to oligonucleotide resolution in the HILIC mode. Keeping in mind the mobile-phase compatibility for LC–MS analysis, Gong and McCullagh (17) investigated such resolutions with other buffers, such as ammonium acetate, bicarbonate, or formate. They achieved a usable separation of an 18-mer oligonucleotide from its 18-mer, single methylated version, with a 100 mM ammonium acetate buffer on a 100 mm × 2.1 mm, 3.5-μm dp HILIC column. From these reports, it appears that HILIC can resolve oligonucleotides up to a 20-mer length (15). This was a poorer, separation performance than achieved with state-of-the-art reversed-phase LC columns (18). It remains to be seen whether HILIC can provide useful selectivity for the separation of phosphorothioate, phosphodiester, or depurinated species, because these are relevant impurities of oligonucleotide drugs that are often unresolved by reversed-phase LC.
Polar Lipids
HILIC has also found applications in lipidomic research, particularly for the characterization of polar lipids (19–21). Because of its nature, HILIC is capable of resolving lipids (phospholipids) according to their polar head group. The lipids are resolved into classes according to their polarity (Figure 1), and the species are separated according to the hydrophobic portion of the lipids (fatty acid chain length), which is typically not achieved (21). On the other hand, the separation of fatty acids of different lengths and degree of unsaturation (number or position of double bonds), is readily achieved in reversed-phase LC. Therefore, the HILIC × reversed-phase 2D LC–MS method is currently being explored for advanced polar lipids analysis (21).


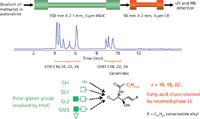
Figure 1: Analysis of polar ceramide lipids. Two columns, HILIC and reversed phase, were connected in series. Lipids were eluted with a methanol gradient. Mobile-phase A: 95% acetonitrile, 5% methanol, 5 mM ammonium formate, and 0.2% formic acid. Mobile-phase B: methanol, 5 mM ammonium formate, and 0.2% formic acid. Ceramides were eluted with a methanol gradient at a flow rate of 250 μL/min. The HILIC first dimension resolves analytes according to their hydrophobic glycan group; the reversed-phase column separates fatty acid chains according to their length while being insensitive to glycan moiety. Identification was performed by MS. (Adapted with permission from reference 21.)
Glycosylation
HILIC shows significant promise in the analysis of protein glycosylation. As discussed in the first part of this series (22), HILIC is a very useful method for the analysis of released glycans, together with fluorescent detection. This approach provides both high resolution and sensitivity, but has limitations when used for site specific glycosylation analysis. Upon cleavage of glycans from proteins, one loses information about the original site heterogeneity. In other words, if two glycosylation sites contain the same glycan, these species are pooled before analysis, and the original level of glycosylation at each site remains unknown. That is why some researchers explored an alternative approach based on proteolysis of the glycoprotein followed by analysis of the resulting glycopeptides (23,24). The selection of methods used for glycoprotein analysis via glycan or glycopeptides routes is shown in Figure 2. The unique peptide "mass tag" attached to the glycans can identify the nature and degree of glycosylation at each site (4,25). A second major advantage of the glycopeptide based approach, is the ability to directly interrogate O-linked glycans, thus removing the need for specific O-linked deglycosylation enzymes, which are not readily available at this time (24,26,27).

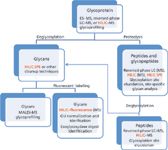
Figure 2: Glycoprotein analysis scheme. When applicable, HILIC technique is highlighted in red.
Glycopeptide analysis utilizes two unique HILIC features. First, the glycosylation (addition of a highly polar moiety) causes a marked enhancement of peptide retention in HILIC (Figure 3). Consequently, the glycosylated peptides are typically resolved from nonglycopeptides present in the protein digest (4,24). Second, the glycopeptides are separated into their glycoforms, including positional isomers of glycans (Figures 3 and 4). This is useful for both ultraviolet (UV) and MS analysis, given the isobaric nature of isomers and potential for in-source fragmentation of labile glycan moieties (4).


Figure 3: HILICâUVâMS analysis of tryptic digests of (a) mAb and (b) human 1-alpha-acid glycoprotein. Glycopeptides are more retained in HILIC (highlighted in blue). The elevated collision energy MS spectrum is shown in the bottom panel for the peak labeled with an asterisk. The glycan moiety is fragmented into characteristic ions, confirming that the selected peak is indeed a glycopeptide. For chromatographic conditions and details see reference 4. Adapted with permission from reference 4.
Glycoproteins
Several groups have adopted HILIC for systematic studies of glycoproteins. Two distinct schools of thought can be observed. Some researchers prefer using specific enzymes for protein digestion such as trypsin, Glu-C, or Lys-C. In such an approach, the proteolysis creates highly specific glycopeptides and, desirably, one peptide per glycosylation site (4,23,24,28–32). These peptides are then analyzed by HILIC-MS and the heterogeneity and nature of glycosylation are revealed. Unfortunately, in some cases the proteolysis creates long peptides with masses of 3–6 kDa, which do not present easy targets for MS analysis. The lack of suitable cleavage sites can also create peptides with two or more glycosylation sites and compromise the method's capacity for site-specific glycosylation analysis (4).


Figure 4: HILIC MRM analysis of tryptic digest of purified murine IgG1 antibody. EEQFNSTFR tryptic peptide glycoforms are resolved in HILIC (for conditions, see reference 4). MRM permits selective detection and quantification of all glycopeptide species, even chromatographically unresolved ones. The quantitation results were comparable with the HILIC-fluorescence method (39). (Adapted with permission from reference 39.)
A second approach uses nonspecific proteolysis with Pronase or Proteinase K enzymes (25–27,33,34). The cleavage creates short, 2–6 amino acid long glycopeptides, which lend themselves well to MS analysis. In addition to labile glycan moiety fragments (most abundant are 204.09, 366.14, and 657.24 Da, see inset in Figure 3b), the collision-induced dissociation (CID) spectrum of a glycopeptide contains signals of the peptide or peptide+GlcNac ions (31–33,35). These both provide information about the glycosylation site. The site identification is typically confirmed by multiple peptides generated by nonspecific enzymatic cuts around the glycosylation site. However, the multitude of signals complicates the HILIC-MS data interpretation (25,26,34) and dilutes the available signal. Low-abundance glycans may not be detected (33).
Besides analysis in the column format, HILIC solid-phase extraction (SPE) methods have been utilized for glycopeptide enrichment from complex peptide samples (Figure 2). This has become a very popular and useful overall approach for improved glycan analysis and glycoprofiling. Micro-SPE tips (30), short HILIC guard columns (29), and a batch SPE approach (28,29) have been described. Although some authors prefer multilectin specific enrichment of glycopeptides or glycoproteins (36), the simple HILIC glycopeptide enrichment has gained in popularity (30,37). One should be aware, though, that some hydrophobic glycopeptides with a short glyco moiety (O-linked peptides), may not be sufficiently resolved from hydrophilic nonglycopeptides. HILIC SPE glycopeptide enrichment is a fast and convenient method, but it does not remove all nonglycosylated peptides from the enriched glycopeptides (4,30).
Several groups have successfully used the HILIC glycopeptide approach for analysis of singly glycosylated proteins such as monoclonal antibodies and IgGs (4,23,29,32). However, the analysis of multiple glycosylated proteins remains a difficult task. Multiple glycoforms of each glycopeptide share the same HILIC separation space, and thus challenge the separation capability of HILIC. Even when MS is used, it is difficult to perform the data interpretation (4). This is especially the case for samples like vaccines that contain multiple viral hemagglutinin isoforms as their immunogenic proteins (35,38). A combination of 2D LC and MS may provide a suitable solution. Reversed phase × HILIC 2D LC relies on the fact that reversed-phase LC resolves glycopeptides by the peptide hydrophobicity, whereas the peptide glycoforms are eluted as a single peak or a closely spaced group of peaks (29). HILIC, on the other hand, can resolve a given glycopeptide into its glycoforms (Figures 3 and 4). Off-line 2D LC with the first dimension consisting of reversed-phase LC and the second dimension of HILIC–UV–MS was evaluated (4). MS offers an alternative to UV detection, as well as a highly desirable sensitivity for the detection of glycopeptides, often present at significantly lower concentrations than other peptides in the digest. The concentrations are lower because only a certain fraction of proteins could be glycosylated and because the glycopeptide signal is split into subspecies as a result of the glycan site microheterogeneity. Minor glycoforms are often present at levels less than 10%. Sensitive triple-quadrupole MS detection, utilizing multiple reaction monitoring (MRM) of glycopeptides, was demonstrated (39), capable of the detection of minor glycoforms (Figure 4). The MRM sensitivity was comparable with the HILIC–fluorescence technique (39,40), and it permits the quantitation of glycoforms that are chromatographically unresolved. It remains to be seen whether this promising approach will be adopted as a workhorse method for glycosylation analysis.
Finally, the complicated task of metabolomic profiling can take advantage of the 10–100× increased sensitivity of HILIC available with LC–MS detection. Also, as can be done with any set of small molecules that differ by polar group composition, one can take advantage of the multimodal mechanisms available to HILIC separations. This quality makes it particularly suited for better selectivity by amplifying these types of differences rather than using reversed-phase LC or other modes of partition chromatography.
Other Applications
HILIC has been described and utilized for more than two decades and while a substantial amount of literature covers the field and there is substantial commercial interest in its applications and usage, most of this has involved, so far as biotechnology is concerned, glycan (glycoprofiling) (22) and glycopeptide analyses (vide supra). Much less of an effort has been described for glycoproteins or larger species (41–54). Very little usage has been made for perhaps the most important player in biotechnology today: recombinant antibodies (MAbs). In general, HILIC has been applied far more for low-molecular-weight species, conventional drugs, forensics, natural products, and virtually any analyte that has properties compatible with HILIC's separation strengths. This means, in general, that the analyte must be fairly hydrophilic, polar, partially ionic, or a combination of all three of these separation properties. It must also be fairly water or alcohol soluble, and less organic miscible, or have a mixture of both aqueous and organic solubilities.
In glycan and glycopeptide separations, it is the carbohydrate portion of these molecules that makes the analytes suitable for HILIC and perhaps less suitable for reversed-phase LC. In the case of glycopeptides, they are suitable for both reversed-phase LC and HILIC approaches because their organic and aqueous solubilities and physicochemical interactions with the mobile phases are approximately equal. This may well be why 2D LC incorporating both HILIC and reversed-phase LC offers such important advantages. In the case of glycoproteins or higher order species (MAbs) this is no longer the case and their organic solubilities usually overshadow aqueous miscibility and physicochemical interactions. Glycoproteins are more organic-like than aqueous, and hence they are much more miscible with organic solvents than with water or alcohols.
Thus, it is no surprise that the vast majority of HILIC applications in the biotechnology areas, so far at least, have been glycans and glycopeptides. This situation may well remain true until we discover how to make HILIC much more glycoprotein friendly than it is at present, perhaps by using multiphase HILIC supports. However, that approach then introduces ion-exchange separation modalities, perhaps more so than HILIC interactions, which then muddies the interpretation of whether the separation mechanism is pure HILIC or if it is mixed mode. In any event, the number of literature reports or application notes wherein HILIC has been used for glycoproteins, or proteins in general, remains far lower than for glycans and glycopeptides. However, there are some very useful and interesting reports, which we will now proceed to relate.
Much of the initial theory and methods for HILIC of peptides or proteins originated with Alpert of PolyLC, often in collaboration with various academic colleagues. In several of their presentations and commercial literature or applications, specific protein results are attributed as arising from such colleagues. For example, in 2006 Carroll and colleagues (53,54) described a number of different types of proteins successfully separated by HILIC methods. Figure 5 provides a very nice chromatogram for a mixture of intact mitochondrial membrane proteins. The sample was an organic extract of bovine heart mitochondria, with a very large variety of different proteins eluted in about 1 h. Because of the nature of the two phases, this is a classic HILIC type separation. Figure 5 also indicates the names of some of the proteins involved, all of which were eluted in 60 min, with the gradient conditions indicated. This particular work is discussed in greater detail below.


Figure 5: HILIC of intact mitochondrial membrane proteins (47). Column: 100 mm à 2.1 mm PolyHydroxyethyl A; gradient: 20 mM ammonium formate (pH 3.7) with 0.5% HFIP; (63% 2-propanol, 22.5% acetonitrile) to 30% 2-propanol. Sample: 2-propanolâacetonitrile (pH 3.7) extract of bovine heart mitochondria. (Reproduced with permission of PolyLC and John Walker, Medical Research Council Dunn Human Nutrition Unit, Cambridge, UK, 2006.)
Another illustration of HILIC protein separations is given in Figure 6, which works well for membrane-type proteins (55). Again, a linear gradient was used. One other, early example of the capabilities of HILIC, for difficult to resolve membrane proteins, is shown in Figure 7, again from PolyLC (56). In this particular application, the same HILIC column was used with somewhat different gradient elution conditions, as given. The identified proteins are indicated in Figure 7. The total time for elution was <40 min. Several other applications of HILIC for different protein types have been described over the years in applications from PolyLC and others (for example, histone H4 acetylation and methylation variants, and cation-exchange HILIC on a PolyCat A column with a gradient of sodium perchlorate in 70% acetonitrile was able to resolve histone H1.5 phosphorylation variants) (57).


Figure 6: HILIC works well for membrane proteins, with removal of SDS, Coomassie Blue, and salts from an electroeluted membrane protein. Column: 200 mm à 4.6 mm, 5-μm dp PolyHydroxyethyl Aspartamide; gradient: 70â0% n-propanol in 50 mM formic acid. (Reproduced with permission from A. Alpert, PolyLC, and P. Jeno, University of Basel, Basel, CH.)
As already suggested, PolyLC probably has the largest number of HILIC-based protein separations of any commercial vendor and these proteins are usually difficult to resolve using other HPLC approaches. Thus, on one of their website pages (58) the company describes several such valuable applications as follows. One such application involves using strong cation-exchange HILIC for lung surfactant proteins and their variants, again using the company's PolySulfoethyl A column, with a mobile-phase gradient combining methylphosphonic acid, sodium perchlorate, acetonitrile (mobile-phase A) and 100 mM sodium perchlorate as mobile-phase B. In another application, phosphorylation variants of histone H1 were separated on a PolyCAT A column using 70% acetonitrile in the mobile phases, and resolution was excellent, with a reversal in the order of elution when compared with 0% acetonitrile. Other histone H1.5 phosphorylation variants were similarly resolved (58,59). HILIC modes were superior to capillary electrophoresis for separation of these particular protein variants.


Figure 7: Top-down proteomics of hard samples, HILIC of mouse brain membrane proteins. Column: PolyHydroxyethyl A ; gradient: 70â40% acetonitrile in Tris-HCl (pH 6.5) with 40 mM HFIP; sample: brain homogenate pellet extracted with HFIP. (Reproduced with permission from A. Alpert and S.D. Garbis, Academy of Athens, ASMS 2005 poster #ThP 563.)
Histone H4, acetylation, and methylation variants were well resolved on a PolyCAT A column, operated in the HILIC mode (58). This particular column has both cation-exchange and HILIC properties, so that hydrophobic interactions can be superimposed on the electrostatic effects. The column was also able to separate variants with the same number of acetylated Lys residues, but differing in the number of methylated Lys residues. The identification of these variants was only possible by using this top-down separation step before digestion of individual resolved variants and bottom-up peptide mapping identification (59). If not fully resolved, they would have been eluted together with the far more abundant, less highly acetylated variants. In such a case, it would have been much more difficult to unambiguously identify both the acetylation and methylation variants (58,59).
In one of its on-line discussions of HILIC (60), PolyLC discussed the general applicability of HILIC for intact proteins: "While water-soluble proteins such as cytoplasmic enzymes do not lend themselves well to HILIC, HILIC works well for proteins that do not normally occur free in aqueous solution, such as membrane proteins and histones."
Let us return now to the scientific literature for some final, described applications of HILIC for proteins of various properties and sizes. In a very recent publication, the authors reviewed past efforts to utilize HILIC in any form for intact proteins (61). They again stated that, other than the original publication by Alpert (1), very little had been published on the purification of proteins by HILIC. The authors nicely review most of the prior literature, which was perhaps two or three previous publications that used HILIC or variants thereof for analyzing proteins. They further speculated as to reasons for this seeming anomaly was that many proteins would be denatured, with concomitant loss of bioactivity, in the presence of the high concentration of organic solvents needed in HILIC. However, many classes of proteins do tolerate such conditions without denaturation. In any event, most HPLC work is directed at the detection and measurement of proteins, not at determining their biological activity. Another perhaps more valid reason may have been that many proteins are not soluble in the high concentrations of organic solvents usually needed for HILIC. However, to some extent, the lack of solubility can be countered by the addition of solubilizing agents such as hexafluoroisopropanol (HFIP) or unbuffered acids to the solvents for HILIC. In any event, the number of successful applications of HILIC for intact proteins has clearly grown in recent years, both academically and commercially.
Tetaz and colleagues (61) used the initial chromatographic conditions described by Carroll and colleagues (54) and then developed a modified method that permitted the chromatography of numerous intact soluble proteins by HILIC (Figure 8). In some instances, it was possible to recover functionally active proteins. Their work described the resolution of two different lipophilic proteins (apoA-I and a truncated version of human apoM, lacking its signal peptide), plus another mitochondrial membrane-associated protein. The work was done on five different commercially available columns. It was further demonstrated that HILIC may be used to separate glycosylated isoforms of intact human apoM (61). In this study, small aliquots of the collected HILIC fractions of interest were directly analyzed by nanoelectrospray MS using quadrupole time-of-flight (TOF) tandem MS. The specific gradient elution HILIC conditions, finally optimized on various columns, are given and are very similar to those reported earlier by Carroll and colleagues (54).

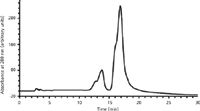
Figure 8: HILIC separation of human apoM (1 mg) on a TSKgel Amide80 column. (Adapted with permission from reference 61.)
These authors were able to demonstrate the successful chromatography of both apoA-I and apoM, an additional class of lipophilic proteins not described before in HILIC (61). However, a more interesting and potentially significant finding was obtained with the separation of glycosylation variants of apoM. This suggested the potential of HILIC for separations never previously attempted or described. Figure 8 shows the HILIC separation of human apoM on a TSK gel Amide80 column (Tosoh Bioscience), with conditions described (61). Identification of the various proteins present in Figure 8 was done by analyzing collected fractions. As shown by the ESI-MS data, the earliest eluted peaks at 13–15 min were nonglycosylated apoM, and the fractions from 16–18 min were different, glycosylated isoforms of apoM. This may have been one of the first reports of an HPLC technique that could separate different glycosylated isoforms of an intact glycoprotein, other than by use of affinity chromatography (lectin, antibody, or boronic acid groups). The HPLC resolution of glycosylation variants of any protein has been a very difficult proposition, although separations of glycosylation variants of peptides have been much easier to realize, as described elsewhere in this column, vide supra.
In a separate, but quite related publication, Carroll and colleagues (54) defined the mitochondrial proteome by measurement of molecular masses of numerous, membrane proteins. As the authors stated,
"The use of this form of chromatography for purifying membrane proteins [together with SDS-PAGE with marker proteins and ESI-MS analysis of the HILIC fractions to confirm molecular masses] is a new development that has allowed some well known, but hitherto poorly characterized proteins to be more thoroughly studied."
Figure 9 illustrates the overall approaches used in this particular study (note that Figure 9a is the same as Figure 5). There are two HILIC chromatograms, showing elution times of the major protein peaks and the nature of the gradient used for elution of each extract. Various protein fractions from each chromatogram were manually collected, transferred to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and run alongside known protein markers of known molecular weights (data not shown). These were then (tentatively) identified by standard Coomassie Blue staining, and the molecular weights were approximately determined by the standard, known molecular weight marker proteins, as usual. Individual SDS-PAGE bands were cut from the plates, destained, denatured, and digested by trypsin to perform peptide mapping ESI-MS and database identification of the original proteins present. Each protein eluted from the HILIC column was also subjected to ESI-MS to determine its correct absolute molecular mass. In the above manner, especially improved by the use of the HILIC protein separation step, it was then possible to identify each of the major proteins present in Figure 9a and 9b (54). This technique was similarly described in proteomics by Berger and colleagues (62,63) and is now commonly termed middle-out proteomics.


Figure 9: Fractionation of hydrophobic proteins by HILIC. Solvent extracts of bovine mitochondria and complex I were made at pH 3.7 and then fractionated by HILIC. Different elution fractions from each chromatogram were then analyzed by SDS-PAGE with known marker standards (54). Elution profiles (solid lines) were monitored by UV absorbance at 225 nm. The column was equilibrated in buffer 1 and analytes were eluted with a gradient (dotted lines) of buffer 2. Flow rates: (a) 0.1 mL/min, (b) 0.05 mL/min. (Reproduced with permission of Proceedings of [US] National Academy of Sciences [PNAS] and the National Academy of Sciences US.)
Conclusions and Possible Future Applications for HILIC in Biotechnology Tomorrow
In summary, when we consider both parts I and II on this subject, and the major applications of HILIC in the literature and commercial materials today, it is clear that the method has become widely accepted, applied, and improved, with more commercially available HILIC columns as well as their accompanying, application notes. This is especially true for the three major classes of applications: glycans, glycopeptides or peptides, and glycoproteins or proteins. Although the first two classes have received the main emphasis in the literature and commercially since about 1990, it is hoped that applications for glycoproteins or proteins and the other groups alluded to above will soon increase and become more widespread. In addition, it should be clear that HILIC may well be an ideal approach for many other types of mono- and oligosaccharides, especially for heparins, glucagons, and more-widespread applications in glycomics (complex oligosaccharides such as glycosaminoglycans). It is also hoped that HILIC will play an increasingly large role in the proteomics and peptidomics areas, especially via the 2D LC–MS formats described above. And finally, it is hoped that HILIC will continue to be found advantageous for glycopeptide isolation in typical, protein-derived peptide mapping, often in combination with reversed-phase LC (2D LC). It is also entirely feasible that the approach will soon show utility for improved resolutions in the HPLC analysis of complex oligonucleotides, as suggested above.
Acknowledgments
The authors acknowledge with gratitude and appreciation the assistance and guidance provided by several colleagues. We acknowledge receipt of numerous HILIC application notes and lecture slides, and a MerckSeQuant Primer on HILIC from Dave Lentz, Knut Irgum, and Tobias Jonsson of SeQuant Merck Millipore. We are especially indebted to Dr. Jonsson for providing us with numerous literature and applications references dealing with HILIC of proteins. And, for his astute comments and observations leading to constructive revisions in some parts of the above.
We acknowledge the use of certain figures from various presentations by Andy Alpert and his applications literature from PolyLC. We acknowledge receipt of another Primer on HILIC from Waters Corporation, as well as the use of several of their application notes on HILIC for biotechnology applications.
Finally, we acknowledge the invaluable technical assistance from Simion Kreimer of Northeastern University, in pulling together many of the final figures used in part II.
References
(1) A.J. Alpert, J. Chromatogr., A 499, 177–196 (1990).
(2) A. Zhu, J. Martosella, and P.T. Duong, Application Notebook, supplement to LCGC North Am., pp. 8–10 (September 2013).
(3) M. Gilar and A. Jaworski, J. Chromatogr., A 1218, 8890–8896 (2011).
(4) M. Gilar, Y.Q. Yu, J. Ahn, H. Xie, H. Wan, W. Ying, and X. Qian, Anal. Biochem. 417, 80–88 (2011).
(5) T. Yoshida, Anal. Chem. 69, 3038–3043 (1997).
(6) C.T. Mant, J.R. Litowski, and R.S. Hodges, J. Chromatogr., A 816, 65–78 (1998).
(7) O.A. Ismaiel and R.G. Jenkins, Drug Test Anal. (2013), doi: 10.1002/dta.1521.
(8) A. D'Attoma, C. Grivel, and S. Heinisch, J. Chromatogr., A 1262, 148–159 (2012).
(9) P.J. Boersema, N. Divecha, A.J. Heck, and S. Mohammed, J. Proteome Res. 6, 937–936 (2007).
(10) S. Di Palma, P.J. Boersema, A.J. Heck, and S. Mohammed, Anal. Chem. 83, 3440–3447 (2011).
(11) D.E. McNulty and R.S. Annan, Mol. Cell. Proteomics 7, 971–980 (2008).
(12) A.J. Alpert, Anal. Chem. 80, 62–76 (2008).
(13) H. Zhang, T. Guo, X. Li, A. Datta, J.E. Park, J. Yang, S.K. Lim, J.P. Tam, and S.K. Sze, Mol. Cell. Proteomics 98, 635–647 (2010).
(14) C.S. Gan, T. Guo, H. Zhang, S.K. Lim, and S.K. Sze, J. Proteome Res. 7, 4869–77 (2008).
(15) P. Holdsvendova, J. Suchankova, M. Buncek, V. Backovska, and P. Coufal, J. Biochem. Biophys. Methods 70, 23–29 (2007).
(16) Q. Li, F. Lynen, J. Wang, H. Li, G. Xu, and P. Sandra, J. Chromatogr., A 1255, 237–243 (2012).
(17) L. Gong and J.S. McCullagh, J. Chromatogr., A 1218, 5480–5486 (2011).
(18) S.M. McCarthy and M. Gilar, Waters Application Note 720003361en 1-8 (2010).
(19) E. Cifkova, M. Holcapek, M. Lisa, M. Ovcacikova, A. Lycka, F. Lynen, and P. Sandra, Anal. Chem. 84, 10064–10070 (2012).
(20) M. Schwalbe-Herrmann, J. Willmann, and D. Leibfritz, J. Chromatogr., A 1217, 5179–5183 (2010).
(21) A. Belenky, A. Kloss, B. Wang, E. Budman, R. Bernasconi, and A. Cohen, "Quantitation of Glycosphingolipids in Rat Tissue Extracts Using 2D-LC-MS/MS Method, Poster P 27.01" presented at the The International Symposium on High Performance Liquid Phase Separations (HPLC), Ghent, Belgium, 2007.
(22) I.S. Krull, A. Rathore, and A. Heckendorf, LCGC North Am. 31(12), 998–1007 (2013).
(23) Y. Takegawa, K. Deguchi, T. Keira, H. Ito, H. Nakagawa, and S. Nishimura, J. Chromatogr., A 1113, 177–181 (2006).
(24) Y. Takegawa, H. Ito, T. Keira, K. Deguchi, H. Nakagawa, and S. Nishimura, J. Sep. Sci. 31, 1585–1593 (2008).
(25) G. Zauner, C.A. Koeleman, A.M. Deelder, and M. Wuhrer, J. Sep. Sci. 33, 903–910 (2010).
(26) G. Zauner, M. Hoffmann, E. Rapp, C.A. Koeleman, I. Dragan, A.M. Deelder, M. Wuhrer, and P.J. Hensbergen, J. Proteome Res. 11, 5804–5814 (2012).
(27) O. Hernandez-Hernandez, R. Lebron-Aguilar, J.E. Quintanilla-Lopez, M.L. Sanz, and F.J. Moreno, Proteomics 10, 3699–3711 (2010).
(28) J. Wohlgemuth, M. Karas, T. Eichhorn, R. Hendriks, and S. Andrecht, Anal. Biochem. 395, 178–188 (2009).
(29) J. Wohlgemuth, M. Karas, W. Jiang, R. Hendriks, and S. Andrecht, J. Sep. Sci. 33, 880–890 (2010).
(30) J. Zhu, F. Wang, R. Chen, K. Cheng, B. Xu, Z. Guo, X. Liang, M. Ye, and H. Zou, Anal. Chem. 84, 5146–5153 (2012).
(31) T. Imre, G. Schlosser, G. Pocsfalvi, R. Siciliano, E. Molnar-Szollosi, T. Kremmer, A. Malorni, and K. Vekey, J. Mass Spectrom. 40, 1472–1483 (2005).
(32) C. Singh, C.G. Zampronio, A.J. Creese, and H.J. Cooper, J. Proteome Res. 11, 4517–4525 (2012).
(33) C.C. Nwosu, R.R. Seipert, J.S. Strum, S.S. Hua, H.J. An, A.M. Zivkovic, B.J. German, and C.B. Lebrilla, J. Proteome Res. 10, 2612–2624 (2011).
(34) M. Wuhrer, C.A. Koeleman, C.H. Hokke, and A.M. Deelder, Anal. Chem. 77, 886–894 (2005).
(35) T.A. Blake, T.L. Williams, J.L. Pirkle, and J.R. Barr, Anal. Chem. 81, 3109–3118 (2009).
(36) C.D. Calvano, C.G. Zambonin, and O.N. Jensen, J. Proteomics 71, 304–317 (2008).
(37) P.H. Jensen, S. Mysling, P. Hojrup, and O.N. Jensen, Methods Mol. Biol. 951, 131–144 (2013).
(38) Y. An and J.F. Cipollo, Anal. Biochem. 415, 67–80 (2011).
(39) H.W. Xie, Y.Q. Yu, J. Ahn, and S.J. Berger, P-135 Mass Spec 2010, Waters poster library, 720003753en.pdf (2010).
(40) P. Dugo, N. Fawzy, F. Cichello, F. Cacciola, P. Donato, and L. Mondello, J. Chromatogr., A 1278, 46–53 (2013).
(41) HILIC Solutions, Separation Science quarterly newsletter, www.sepscience.com (2013).
(42) M.R. Gama, R.G. da Costa Silva, C.H. Collins, and C.B.G. Bottoli, Trends Anal. Chem. 37, 48 (2012).
(43) E.S. Grumbach and K.J. Fountain, "Comprehensive Guide to HILIC, Hydrophilic Interaction Chromatography," Waters Corporation, Milford, MA, 2010.
(44) www.wikipedia.org/wiki/Hydrophilic_interaction_chromatography.
(45) P.G. Wang and W. He, Hydrophilic Interaction Liquid Chromatography (HILIC) and Advanced Applications, Chromatographic Science Series (CRC Press, Taylor & Francis Group, Boca Raton, Florida, 2011).
(46) B.A. Olsen and B.W. Pack, Hydrophilic Interaction Chromatography: A Guide for Practitioners, Chemical Analysis Series (John Wiley & Sons, Hoboken, New Jersey, 2013).
(47) P. Hemstrom and K. Irgum, J. Sep. Sci. 29(12), 1784–1821 (2006).
(48) T. Jonsson, N.P. Dinh, and K. Irgum, "Retention and Selectivity for Hydrophilic Interaction Liquid Chromatography (HILIC) Columns: Multivariate Modeling and Correlation with Physiochemical Properties," presented at the 60th ASMS Conference on Mass Spectrometry and Allied Topics, Vancouver, Canada, 2012.
(49) N.P. Dinh, T. Jonsson, and K. Irgum, J. Chrom., A 1218(35), 5880–5891 (2011).
(50) Merck SeQuant, Box 7956, SE-90719, Umea, Sweden, USA: EMD Millipore, Billerica, MA.
(51) P. Hemstrom, T. Jonsson, W. Jiang, and P. Appelblad, Merck SeQuant AB, Umea, Sweden, Application Note (2013); www.merckmillipore.com/chromatography.
(52) PolyLC, Columbia, Maryland.
(53) A. Alpert, "Hydrophilic Interaction Chromatography (HILIC) With and Without Electrostatic Effects: Amino Acids to Proteins," presented at Conference on Small Molecule Science (CoSMoS), Boston, Massachusetts, 2009, Figure 14.
(54) J. Carroll, I.M. Fearnley, and J.E. Walker, PNAS 103(44), 16170–16175 (2006).
(55) A. Alpert, PolyLC, Columbia, Maryland, "Hydrophilic Interaction Chromatography for Membrane Proteins," Data/slide courtesy of P. Jeno, University of Basel, Basel, Switzerland.
(56) A. Alpert, PolyLC, Columbia, Maryland, "Top-Down Proteomics of Hard Samples, HILIC of Mouse Brain Membrane Proteins," Data/slide courtesy of S.D. Garbis et al., Academy of Athens, ASMS 2005 poster #ThP 563.
(57) PolyLC, Columbia, Maryland, Application notes dealing with HILIC or cation-exchange HILIC of protein variants.
(58) http://www.polylc.com/IEX.v.2.htm.
(59) B. Sarg, W. Helliger, H. Talasz, B. Forg, and H.H. Lindner, J. Biol. Chem. 281, 6573-6580 (2006).
(60) PolyLC, Columbia, Maryland, http://www.polylc.com/HILIC.v.2.htm, August 31, 2013.
(61) T. Tetaz, S. Detzner, A. Friedlin, B. Molitor, and J-L. Mary, J. Chromatogr., A 1218, 5892–5896 (2011).
(62) S.J. Berger, K.M. Millea, I.S. Krull, and S.A. Cohen, in Separations in Proteomics, G. Smejkal and A. Lazarev, Eds. (Taylor and Frances, Publishers, New York, New York, 2006), Chapter 21.
(63) K.M. Millea, I.S. Krull, S.A. Cohen, J.C. Gelber, and S.J. Berger, J. Proteome Research 5(1), 135–146 (2006).
Dr. Martin Gilar is a principal investigator in the Core Research group at Waters Corporation in Milford, Massachusetts. He has more than 20 years of experience with separation of oligonucleotides, peptides, glycans, and proteins. He has authored over 40 research papers.

Dr. Martin Gilar
Dr. Heckendorf has 40 years of chromatographic experience in natural products and biological separations. His company, The Nest Group ("Where Little Things Begin to Grow"), supplies and supports new, and sometimes unconventional chromatographic materials or approaches. He is currently investigating chromatographic limits on micro-SPE tip column methods and is developing data on trapping efficiency for LC–MS applications.


Dr. Heckendorf
Ira S. Krull is Professor Emeritus of Chemistry and Chemical Biology at Northeastern University, Boston, Massachusetts, and a member of LCGC's editorial advisory board.

Ira S. Krull
Anurag S. Rathore is a professor in the Department of Chemical Engineering at the Indian Institute of Technology in Delhi, India.

Anurag S. Rathore















Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.


Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.




