Column Dead Time as a Diagnostic Tool
LCGC North America
What good is that big, ugly peak at the beginning of the chromatogram?
What good is that big, ugly peak at the beginning of the chromatogram?
Often considered a necessary evil, the first peak in a chromatogram can be a useful diagnostic tool for troubleshooting liquid chromatographic (LC) separations. Most people I encounter refer to this as the column dead time peak, abbreviated t0. However, it has a wide variety of other names: junk peak, garbage peak, solvent front, or hold-up time, with tM as the most common alternative abbreviation. This represents the time it takes something to go through the LC column that does not interact with the column. A corresponding dead volume (or hold-up volume), VM, is the volume of mobile phase inside the column. This volume comprises both the volume of mobile phase between the packing particles (the interstitial volume) and the volume within the particles (the pore volume). We'll see that t0 can be a useful diagnostic tool to identify potential problems with an LC method.
Measuring t0
If we want to use the column dead time as a tool, we need to be able to identify it. Most LC detectors will generate a peak at t0, the most obvious exception being the mass spectrometric detector (liquid chromatography–mass spectrometry [LC–MS]). Therefore, the chromatogram usually has a peak similar to the first baseline disturbance in Figure 1. If the sample is very clean and has minimal unretained material, a small baseline disturbance as shown in Figure 1a may appear. More commonly, there is sufficient unretained material to generate a large, off-scale peak (Figure 1b). Although there are more-exact measurement techniques for t0, such as injection of D2O, most of us just use the retention time of the peak. I prefer to pick a measurement that is easy to reproduce, because most of the time an estimate of t0 is sufficient. For Figure 1a, this is the point the disturbance crosses the baseline, noted by the arrow. Because a large unretained peak usually is off scale so that the top of the peak may be inconvenient to locate, I usually pick the point where the peak rises from the baseline (arrow in Figure 1b). Of course the retention time reported by the data system is another convenient measurement of the dead time.


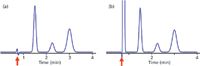
Figure 1: Examples of t0 peaks (arrows). (a) Chromatogram with little unretained material; (b) large peak normally observed at t0.
To confirm a measured value of t0 or to determine it if there is no corresponding disturbance in the baseline, as with LC–MS, we can estimate the column dead volume, VM, and convert it to t0. If you are using a 4.6-mm i.d. column, VM can be estimated as follows:


where VM is in milliliters and L is in millimeters. Thus, for a 150 mm × 4.6 mm column, VM = 0.01 × 150 mm = 1.5 mL. For columns of other internal diameters, you can use
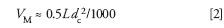
where dc is the column internal diameter in millimeters. For a 50 mm × 2.1 mm column, VM ≈ 0.5 × 50 × 2.12/1000 = 0.11 mL. Either of these estimates is good to within approximately ±10% for columns packed with totally porous particles. These estimates are based on the assumption that VM represents ~65% of the volume of an empty column and that about half of this volume is inside the particles and half is between the particles.
The column dead time is simply the column volume divided by the flow rate, F (in milliliters per minute):

Using t0 as a Diagnostic Tool
I regularly use t0 to help diagnose problems submitted to me by readers. Here are some of the ways this can be useful.
Verify the Unretained Peak
I find that it is useful to check to be sure that the presumed t0 peak is in the right place. For this, simply calculate t0 using equation 1 or 2 and 3, then compare this to the observed peak in the chromatogram. For example, if the chromatogram of Figure 1b was obtained with a 150 mm × 4.6 mm column operated at 2 mL/min, t0 ≈ 1.5 mL/2 mL/min = 0.75 min. This agrees with the observed peak at approximately the same retention time. If the calculated and measured values of t0 differ by more than ~20%, it is advisable to try to figure out why. Several possibilities are discussed below.
t0 Larger Than Expected
If t0 is larger than expected, the most likely cause is a flow-related problem. For isocratic methods, the retention time of retained peaks should change by the same proportion as t0 when the flow rate is changed. If, for example, the above case had an observed t0 of 1.0 min, the retained peaks should increase by 1.0/0.75 = 1.33-fold. If this is confirmed, check for flow-related problems. Larger than expected values of t0 indicate a drop in the flow rate. Always check the most obvious case first — is the flow rate set properly? It also should be obvious that historical retention data should be consulted to be sure an abnormality in t0 really exists.
Assuming that the flow rate is set correctly, the most likely causes of a flow rate problem are leaks, air bubbles, and problems with the check valves or pump seals. A secondary symptom may be low pressure, depending on how far off t0 is. If leaks are not obvious, I would open the pump purge valve and run 5 mL or so of solvent to waste from each flow channel in use. Thus, if you are using a two-pump high-pressure mixing system, purge both pumps; if it is a low-pressure mixing system, purge each solvent line. This should remove any bubbles from the system. If the problem persists, carefully check each fitting in the pressurized flow stream for leaks. Sometimes the fine point of a twisted laboratory wipe or facial tissue can be used to probe the fittings for possible leaks. If leaks in the flow stream are not found, a pump problem is most likely.
If you are using acetonitrile as one of the solvents and the pump does not have active check valves, it is possible for the inlet check valves to stick. This is from the formation of polymers on the surface of the check-valve seat, and usually can be corrected by sonicating the check valves for a few minutes in methanol. A more detailed discussion of this can be found in an earlier "LC Troubleshooting" column (1). The outlet check valves also can leak if they become contaminated. If you have a pump with outlet check valves, sonicating them may help. If you choose to sonicate the check valves, be careful that you know how they are assembled, in case they come apart in the process. Worn pump seals also can leak, resulting in a lower than expected flow rate. Check the maintenance log for the pump. If the seals haven't been replaced in the past year, I would suggest replacing them. If the seals are newer, you can inspect the pump more closely for possible signs of leaking. Most pumps have a hole or drain tube below the pump head behind the check valves, where any leakage from the seals will exit. Look for signs of leakage, such as visible liquid or white deposits of buffer residue. Replace the pump seals if there is any question of its integrity.
t0 Smaller Than Expected
If the observed t0 peak comes out earlier than expected, one of two possibilities exists. The easier to address is a mistake in the flow rate setting so that the flow rate is too high. Although I suppose it is possible, I have never heard of a pump or controller software failure that resulted in excessive flow rates, so operator error is the most likely source of a flow-related problem.
With the most common forms of LC (reversed phase, normal phase, ion exchange, ion pairing, and so forth), t0 should be the first disturbance in the chromatogram. If something is eluted earlier than the expected retention of t0, the compounds may be excluded from the pores of the packing material. The exception to this is in size-exclusion chromatography, where everything should come out before t0. If a molecule is restricted from entering the packing pores, it only has access to the volume of column between the particles (the interstitial volume), which I mentioned at the beginning was approximately half of the total solvent volume, or 30–35% of the column volume if the total dead volume is ~65%. I've seen the interstitial volume quoted as 40% of the column volume (2), but in either case, a significant portion of the dead volume is inside the particles. If sample molecules are restricted from entering the pores of the packing, they will be eluted before t0. Such peaks may be confused with the true t0 peak, because we are used to assigning the first peak in the chromatogram as t0.
Two common causes of restricted access to the pores exist. The most obvious is that the molecules are too large to enter the pores. A rule of thumb is that the pore diameter should be 3–4 times the hydrodynamic radius of the molecule. Most analytical columns have pores in the 8–12 nm (80–120 Å) range, which will accommodate molecules of up to ~10,000 Da. Above this size, for example with proteins, larger-pore columns, with 30–40 nm pores, are used. Thus, if your sample is a pharmaceutical product, although the analyte of interest may be <1000 Da, the formulation may contain polymers or other excipients in excess of 10,000 Da that may be excluded from the pores. In some cases, dimers or other aggregations of sample molecules can result in a material that is stable enough to chromatograph, but too large to enter the pores. Any of these large molecules may be eluted before the column dead volume.
A second cause of restricted access to the pores is that chemical repulsion between a sample molecule and the pore may exist. An example of this is illustrated in Figure 2 (3). In Figure 2a, a sample of adrenaline (base), benzyl alcohol (neutral), and naphthalene sulfonate (acid) is separated on a C18 column with a methanol–buffer mobile phase at pH 6. Adrenaline has a pKa of 8.55, so it will be fully ionized under these conditions, and naphthalene sulfonate has a pKa of <1, so it will also be ionized. Adrenaline is unretained because in its charged form, it is very polar and not retained. On the other hand, naphthalene sulfate has sufficient nonpolar nature that it is well retained, even though it is ionized. With the addition of 14 mM octane sulfate as an ion-pairing reagent, the results of Figure 2b are obtained. Here, the ion-pairing reagent is assumed to be immobilized on the surface of the C18 stationary phase, creating an in situ ion-exchange surface with a negative charge that is used to retain the positively charged adrenaline. However, although the pH of the mobile phase was not changed, you can see that the naphthalene sulfonate is now unretained. This is because the pores now contain a net negative charge that repels the negatively charged naphthalene sulfonate. You can imagine a similar situation where a pore with a net charge would repel a sample molecule of opposite charge, resulting in exclusion from the particles and elution before the column dead time. This phenomenon, called ion exclusion, occasionally is observed in ion-exchange chromatography. Any chemical change in the pore surface that repels sample molecules will have the same result.

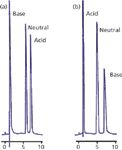
Figure 2: Illustration of exclusion of a charged molecule from packing pores. (a) No ion pairing reagent present, charged base is poorly retained, acid is retained; (b) with ion pairing reagent, charge on particle surface attracts base, increasing its retention, but repels the acid, excluding it from the pores. See text for details. Adapted from reference 3.
Using t0 to Check for "Good" Chromatography
Another use I make of t0 is to check for the quality of the separation, especially when readers submit problem chromatograms for me to diagnose. For isocratic separations (those with a constant mobile phase composition), the retention factor, k, is calculated as follows:

where tR is the retention time of the peak of interest. The retention factor is a measure of the distribution of the sample between the stationary phase and the mobile phase. As an indicator of chromatographic quality, I like to see 1 < k < 20, or better 2 < k < 10, as has been discussed in past "LC Troubleshooting" columns (for example, reference 4). If k < 1 is observed, the peak is likely to have poor retention-time reproducibility and more likely than strongly retained compounds to have interferences from the tail of the t0 peak. The retention factor can be estimated by using t0 as the unit of measure for retention and measuring retention beginning at the observed value of t0. For the chromatogram of Figure 1b, this is done by dividing the baseline up in units of t0 instead of minutes. The first peak is eluted a little over 1 t0 unit past t0, so it has a k value of a little more than 1. Similarly, the second and third peaks have k values of a bit more than 2 and 3, respectively. Although equation 4 does not apply to gradient elution, the general principle of keeping the first peak away from t0 to avoid interferences still holds. From this standpoint, I like to see the first peak of interest in a gradient come off the column at least 1 t0 unit past t0.
Conclusions
Although at first glance, the solvent peak at the beginning of the chromatogram has no value, it can be a useful tool to help diagnose problems with the chromatogram. We can compare column dead time estimates with the observed retention time of the t0 peak and get an idea of what might be going wrong. When the first peak comes out after the expected retention time, the problem is usually flow-related and most commonly caused by a leak. Peaks that are eluted before the expected retention t0 time are most likely excluded from the pores of the column, because they are too large or are repelled from the pores. So we can see that nothing (t0) really is a useful diagnostic tool.
References
(1) J.W. Dolan, LCGC North Am. 26(6), 532–538 (2008).
(2) U.D. Neue, HPLC Columns (Wiley-VCH New York, 1997), p. 53.
(3) J.H. Knox and R.A. Hartwick, J. Chromatogr. 204, 3–21 (1981).
(4) J.W. Dolan, LCGC North Am. 25(7), 704–709 (2007).
John W. Dolan "LC Troubleshooting" Editor John Dolan has been writing "LC Troubleshooting" for LCGC for more than 30 years. One of the industry's most respected professionals, John is currently the Vice President of and a principal instructor for LC Resources, Walnut Creek, California. He is also a member of LCGC's editorial advisory board. Direct correspondence about this column via e-mail to John.Dolan@LCResources.com

John W. Dolan









Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).




Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.

