Extraction and Preconcentration of Residues and Contaminants in Food Samples Using Membrane Techniques
LCGC North America


Guest authors from South Africa review the application of membranes in the extraction, preconcentration, and separation of various contaminants in food.
Over the last few decades there has been tremendous advancement in the field of sample preparation methods, going hand in hand with the developments of state-of-the-art analytical instruments (1–3). The driving force behind the rapid development in the methods for sample preparation has been the need to replace traditional sample preparation methods, which involved many handling steps and, hence, was laborious, time-consuming, and uneconomical in terms of their high demands for consumables, for example, large volumes of solvents (4,5). More recently, an interest in the invention of new, nontraditional sample preparation methods that negate these disadvantages have come about (6,7). Many nontraditional sample preparation methods have been shown to be very selective and efficient in terms of the selective extraction, clean up, and enrichment processes (8).



Ronald E. Majors
The modern sample preparation methods can be classified based upon whether the extraction is exhaustive or nonexhaustive (9,10). A category of nontraditional extraction methods that take place at either exhaustive steady state or nonexhaustive state involving the use of membranes include supported liquid membrane extraction (SLM); microporous membrane liquid–liquid extraction (MMLLE); polymeric membrane extraction (PME); and membrane extraction with sorbent interface (MESI). In this review, we will discuss the various membrane techniques that have been applied in the selective extraction and preconcentration of contaminants in foods. The main attractive feature of membrane techniques in extraction of food samples is that macromolecules such as long chain proteins and carbohydrates are excluded as they cannot pass through the pores of the membrane (Figure 1). Large molecules also have slow mass transfer across the membrane. Selectivity can be finetuned further by optimization of the various conditions such as the donor and acceptor pH (or composition). For liquid membranes, a selective carrier can be incorporated in the organic liquid impregnated in the pores of the membrane increasing the selectivity.

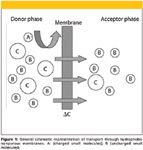
Figure 1
Principles of Membrane Extraction
Both porous and nonporous liquid membranes have been used for extraction and separation of a variety of contaminants in different types of food samples. The difference between the porous and nonporous liquid membranes in terms of their ability to separate mixtures lies from the fact that the porous membrane functions as a boundary separating two solutions, one the receiving phase. The nonporous membranes however, play a role as a selective barrier that governs the selective passage of one analyte species contained in one phase to the other (11). Further, in porous membrane systems, the transport mechanisms, the driving force for mass transfer and the mode of selectivity, are solely dependent upon the partitioning property of the analyte between the contacting phase, which can be liquid–liquid or gas–liquid (12,13). Another important feature with porous membrane is that, the membrane does not have any significant influence as far as selectivity is concerned, though the pore size can exert influence on the selectivity of some specific solutes in some instances (12–15). On the other hand, nonporous membranes normally show the resistance problem for the mass transfer across the membrane, which can affect the mass transfer kinetics negatively (14). In recent reviews of the advances in membrane techniques, various membrane configurations have been detailed (16).
Dialysis: Dialysis and its variant techniques such as microdialysis and electrodialysis are porous membrane techniques widely used in analytical processes (17–21). In typical operations, both the aqueous phases separated by the porous membrane are flowing. Target analytes diffuse from the donor phase to the acceptor phase by a concentration gradient. Dialysis, unlike the nonporous membrane techniques, is not characterized by analyte enrichment; instead there is only partial clean up due to lack of a discrimination mechanism between other nonanalyte molecules that might have the same size as the analyte molecules (22,23). For this reason, dialysis is not regarded as an extraction technique per se, and often an additional sample clean-up step is included in the process. However, it has been applied widely to food samples because macromolecules are excluded from passing through the pores of the membrane (24–26).
Liquid membrane extraction techniques: In liquid membrane techniques, the permeability of any molecule through the membrane is governed by the extent of its solubility to diffuse through the liquid held in the porous membrane and the concentrations of analyte species in the feed and permeate (27). The theory and mathematics of membrane extraction techniques has been well worked out (27,28). The extraction efficiency (E) is defined as the fraction of analyte extracted from the donor phase into the acceptor phase. This is an important parameter commonly measured in membrane technique applications. It is a measure of the rate of mass transfer through the membrane, which is constant at specified extraction time, flow rate, phase composition, temperature, and ionic strength (29,30). The concentration enrichment factor En is a ratio of the concentration found in the acceptor phase to that in the original sample. It is used to estimate the detection limit of the membrane-extraction technique. The volume ratio of the acceptor phase to that of the sample influences the enrichment factor. Usually, acceptor volumes are kept small in the range of a few milliliters to microliters (29).
Supported liquid membrane (SLM): SLM extraction involves a three-phase system (31). An aqueous sample phase (feed–donor) is separated from an aqueous receiver (acceptor) phase by a layer of organic solvent impregnated in the porous membrane. To enhance extractability and selectivity of solutes in SLM, the conditions in the aqueous sample phase normally are adjusted so that the analyte of interest is forward-extracted out of the sample phase into the organic phase and back-extracted out of the organic phase into the receiver phase, in a concerted fashion (29,32,33). In typical operations, the donor is flowing while the acceptor phase is stagnant, thus resulting in high enrichment factors because large sample volume can be extracted. The SLMs can sometimes incorporate a chemical extractant or a carrier to enhance the process of selective transport of analyte components across the membrane interface (29,33). SLM generally is the most selective membrane technique. Ideally, only compounds belonging to the same family should be extracted at a time.
Microporous membrane liquid–liquid extraction (MMLLE): MMLLE is a two-phase technique that involves an aqueous phase and an organic phase. The organic phase is impregnated in the porous membrane and is also part of the acceptor phase.
Selectivity in a two-phase system is based upon the partitioning of the target analytes into the liquid membrane and on the membrane pore size that excludes bigger molecules. A two-phase system is therefore still suitable to extract target analytes from food samples. With a stagnant acceptor phase, the amount of analyte extracted into the organic acceptor phase is limited by its partitioning coefficient into the organic liquid. This system is more applicable for the extraction of nonpolar organic compounds (34). Initially, the flat sheet module was used commonly. However, recently, more applications are using the hollow fiber as the module of choice because phase breakthrough is almost eliminated because no pumping is required as the sample is stirred often. Hollow-fiber modules are also simple and cheap, and high enrichment factors are obtained. Memory effects are also not a problem because of the small thickness of hollow fibers. One limitation is that unlike in the flat sheet module, automation or online extraction is not easy except in the form of the extraction syringe (35), hollow-fiber liquid-phase microextraction (36), and fiber-in-tube liquid-phase microextraction (37) devices.
Hollow-fiber supported liquid membrane (HFSLM) microextraction technique: In hollow-fiber microextraction, a porous polypropylene hollow-fiber strand is used and a very small volume of acceptor solution (in the microliter range) is added to it. The filled hollow fiber is then exposed to an organic liquid to impregnate the pores, and it is then placed in an aqueous sample where extraction will proceed. Hollow-fiber microextraction can be carried out in either a two-phase (MMLLE) or three-phase (SLM) process depending upon the analyte being extracted. The principles of the extraction process that determines the selectivity are therefore similar to those described earlier. The application of the hollow-fiber technique in the extraction and separation of food contaminants has not been reported widely (38–40). However, it is now seen as the module of choice in liquid membrane extractions.
Polymeric membrane (silicone rubber) extraction techniques: A polymeric solid membrane (usually silicone rubber) is used to partition and separate the feed (donor) and stripping (acceptor) phases (41). Selectivity is based upon the differences in the partition coefficients of the analyte and interfering matrices into the silicone rubber, as in MMLLE (41). Conditions of the donor and acceptor phases also can be adjusted, as in the SLM extraction technique (41). Various phase combinations can, thus, be realized, such as aqueous (42) and organic (41). Phase breakthrough is eliminated. Silicone hollow fibers also are available, which can be used as hollow-fiber liquid-phase microextraction (43). The main drawback of PME is that it does not allow additions of chemical extractants (41).
Membrane Extraction of Food Samples
Application of dialysis techniques in food analysis: Methods involving microdialysis have been developed and applied for the extraction and preconcentration of contaminants in foods. Microdialysis has been applied in the extraction of fluoroquinone in chicken liver and muscle (24). In this work, the isolation of the quinolones sarafloxacin, oxolinic acid, and flumequine from fortified chicken liver tissues and sarafloxacin in cured chicken liver tissues was achieved using both liquid–liquid extraction and aqueous on-line microdialysis. The overall recoveries of sarafloxacin, oxolinic acid, and flumequine from samples fortified over a concentration range of 1–100 μg/L were 94, 97, and 87%, and the overall interassay variability was reported to be 4.2, 4.1, and 3.6%, respectively for the three compounds. Chromatograms obtained demonstrate the selectivity of the developed microdialysis method (Figure 2). No other unwanted peaks were coextracted along with the target analytes.

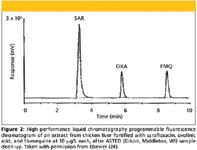
Figure 2
More application of microdialysis has been reported in the determination of lactulose in milk (25). The use of a microdialysis probe as the sampling system for lactulose enabled the direct measurement of lactulose in milk samples without pretreatment in the range between 1 × 10–5 - 5 × 10–3 mol/L. Also, in another application, microdialysis has been used to enhance the sensitivity of a lactate biosensor that was used to determine L-lactate in milk and yogurt (26). When this method was X injection analysis system, a sensitivity of 300 ± 10 nA mmol/L was reported, with a linear response up to 1 mmol/L and detection limits in the low micromolar range. When microdialysis membrane-based sampler was introduced, the sensitivity was found to be 7.9 ± 0.2 mmol/L and it extended the linear range to up to 50 mmol/L lactate.
Nonporous Membrane Extraction Techniques to Food Samples
Supported liquid membrane extraction: There have been several reports on the use of the SLM technique in the extraction of contaminants and/or separation of chemical components in foods and food industry in general. Apart from extraction of aqueous food samples, the SLM extraction principle also can be extended to solid or semisolid samples by incorporating a donor channel unit that permits close contact between the sample and the membrane. Using such a configuration, it has been possible to extract and quantify vanillin in food samples including vanilla sugar, chocolate-coated biscuits, and chocolate (44). In the report by Luque and colleagues, they used a porous PTFE membrane impregnated with di-n-hexyl ether, which forms a barrier between two aqueous phases. Vanillin was then extracted from a donor phase into the hydrophobic membrane and then back extracted into the acceptor. The limit of detection reported was 44 μg/mL.
Another use of SLM in the extraction of caffeine in tea leaves was reported by Zougaph and colleagues (45). In this method, caffeine was extracted from the donor phase through the liquid membrane formed by a mixture of n-undecane and hexyl ether into the acidic acceptor stream. Caffeine was quantified by using a quartz crystal modified with a molecularly imprinted polymer (MIP). The method was reported to have a linear range between 10 and 1000 ng/mL, and a relative standard deviation of ±5% at 50 ng/mL. Msagati and Nindi have reported the application of supported liquid membranes in the extraction of antibiotic and veterinary drug residues in foods of animal origin. They reported the extraction and enrichment of benzimidazole anthelmintics in milk and animal tissues (46,47); extraction and enrichment of sufonamides in milk and animal tissues (48); monitoring of aminoglycosides in milk (49); extraction of penicillin and its related compounds (50), as well as macrolide antibiotics (51). Selective extraction of hormonal residues in food samples has been reported by Msagati and Nindi as well (52,53).
In another report, Miyako and others (54) developed an enantioselective separation method for optically active compounds (S)-ibuprofen and L-phenylalanine from their racemic mixtures using a supported liquid membrane that incorporated a surfactant. The surfactant–enzyme complex that was formed solubilized in the thin organic membrane and effectively catalyzed the esterification reaction in the thin film and another enzyme was used as an ester hydrolysis catalyst in the receiving phase. The (S)-isomer was esterified selectively by the surfactant-enzyme complex at the interface between the donor phase and the liquid membrane. The ethyl ester of (S)-isomer that was produced partitioned into the organic phase of the liquid membrane and diffused across to the acceptor side due to its concentration gradient. At the acceptor–SLM interface, another enzyme catalyzes the ester hydrolysis to produce the initial (S)-isomer and ethanol, which are water-soluble. Thus, the (S)-isomer is transported selectively to the receiving phase through the SLM, based upon the enantioselectivity of the enzymes. A method involving SLM for separation of enantiomers of N-protected amino acid derivatives has been reported (39). In this work, racemic N-protected amino acid derivatives were separated using carbamoylized quinine and quinidine derivatives as carrier, which proved to be very selective. The SLM configuration used by the Maximini group consisted of two polysulfone hollow-fiber membrane modules immobilized with adamantyl-carbamoyl-11-octadecylthioether-quinine (module 1) and adamantyl-carbamoyl-11-octadecylthioether-quinidine (module 2) liquid membranes that was dissolved in 1-decanole–pentadecane in the pores of the hollow fibers in both modules. Using this SLM system, they reported that the compounds were separated with degree of purity of 99% and also in large quantities.
Another separation method involving the use of liquid membranes was reported in which a hollow-fiber liquid membrane was used for the enantioseparation of racemic α-cyclohexyl-mandelic acid that employed copper (II) N-dodecyl-(L)-hydroxyproline (CuN2) as a chiral carrier (38). More application of SLM has been reported in the extraction of biogenic amines in wine using a hollow-fiber module (55). In this report, di-2-ethylhexyl phosphoric acid was used as an anionic carrier incorporated into the membrane phase. In another report, Jung and others (56) developed an SLM extraction for identification of phenolic compounds in the nutrient solution of closed hydroponic growing systems for tomato. In this set-up, flat sheet modules were used and extraction was performed in the field, thereby enabling to monitor the release of such compounds. Figure 3 shows the selectivity of the developed SLM method for extraction of phenolic compounds from nutrient solution. Despite the nutrient solution having high amounts of dissolved organic matter, a clean chromatogram was obtained after SLM extraction.

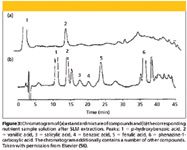
Figure 3
Many applications of the supported liquid membrane extraction technique has been applied to organic compounds in food samples, as cited previously, but there have been few applications for metal ions. This is due to metal ions bound to other components making it difficult to extract the metal across the membrane selectively, even with a suitable carrier. Soko and others (57) attempted to develop a supported liquid membrane extraction method for extraction and preconcentration of manganese(II) from biological fluids (water, milk and blood serum). Di-2-ethylhexyl phosphoric acid (DEHPA) with kerosene as diluent was used as a carrier. The membrane was modified with tri-n-octylphosphine oxide (TOPO) to increase its polarity. Scanning electron microscope images of the membranes revealed that some matrix compounds were deposited on the surface, thus, limiting the extraction process (57).
Polymeric membranes: The use of polymeric membrane techniques has been reported widely, such as in the entrapments of a cysteine protease hydrolase enzyme called papain (58). In this work, pectin extracted from residues of passion fruit (Passiflora edulis, maracuya) was used as a membrane support for immobilization of the enzyme.
Other applications of silicone membrane have been seen in the separation of phenols from crude oils (59). According to their method, phenols were separated from the crude oil matrix using a silicone membrane in a device coupled directly to the chromatographic system. Vitamin E has been determined in butter samples after dissolution of the butter in a micellar medium. Following on-line saponification, the nutrient was enriched across a silicone membrane and taken up in acetonitrile before liquid chromatographic analysis using an electrochemical detector (60). Continuous extraction of vitamin E isomers from vegetable oils also has been reported (61). In this work, samples of thirteen commercially available oils of olive, sunflower, corn, and seed mixtures were analyzed using 2,2,5,7,8-pentamethyl-6-chromanol as an internal standard after they were dissolved in Triton X-114 in the presence of methanol–hexane. The results were validated by analyzing BCR reference material with a certified content of *-tocopherol (margarine CRM 122).
Another membrane separation device has been coupled to a liquid chromatograph for the enrichment of pesticide multiresidues from egg extracts generated via Soxhlet extraction (62). Their report showed that transport through the membrane was related to the octanol–water partition coefficients (log Pow) and to the solubilities of the analytes in the donor and acceptor solutions (hexane and water, respectively). The developed PME method showed good selectivity towards the target analytes as blank chromatogram showed no other unwanted peaks (Figure 4).

Figure 4
Microporous membrane liquid–liquid extraction (MMLLE): The use of MMLLE in the extraction and separation of contaminants in foods also has been reported. Hyötyläinen and others (63) have reported the use of MMLLE in the determination of pesticide residues in wines. The MMLLE developed provided efficient and selective extraction of pesticide residues in wines with enrichment factors in the range 3–13 and was found to be linear over a wide range of concentrations. The limit of quantification obtained was 0.006 mg/L for all the analytes except for one of the analytes known as iprodione, which was found to be 0.37 mg/L. Figure 5 demonstrates the selectivity of the developed method toward target compounds from wine sample.


Figure 5
Lambropoulou and colleagues (64) have recently written an extensive review on the use of MMLLE in the analysis of a wide variety of pesticide classes including insecticides, herbicides, fungicides, antifouling compounds, and phenolic compounds. In another report, Carabias-Martinez and colleagues (41) have employed MMLLE in the extraction of triazine herbicides in cooking oil. MMLLE also has been applied to human breast milk (65) and bovine milk (15) but centrifugation of the samples before extraction was necessary to improve analyte extractability. In these reports, low recoveries were reported due to strong analyte interaction with the matrix.
Challenges in the Applications of Membranes to Sample Preparation of Food Substances
A number of challenges still exist in applications of membranes to food substances. In some cases, the selectivity obtained might not be enough. This is common in two-phase systems, in which the aqueous sample is extracted into the organic phase. This is because selectivity for small molecules is based upon differences on the partition coefficients into the membrane between the analytes and interfering compounds. Stability of liquid membranes in some cases can be problematic, though this depends upon the hydrophobicity of impregnated organic liquid and configuration of the module. This can result in high relative standard deviation values. Instability of liquid membranes also poses a challenge to online extraction because high pressures are generated during pumping of solvents. Another challenge is on membrane biofouling due to adsorption of large molecules in food samples, thus blocking the membrane pores.
Conclusions and Future Perspective
The use of membranes in extraction of contaminants in food samples is getting more attention. The future trend is likely to be focused on using hollow-fiber modules as they are seen as simple, cheap, and offering high enrichment factors as well finding ways for online extraction. Other possible trends will be to combine hollow-fiber extraction with selective sorbents such as molecularly imprinted polymers as well modification of hollow fibers to make them more selective. The latter will require incorporation of the selective functional groups within the hollow fibers. Silicone-based hollow fibers could prove more versatile when applied to food samples because phase breakthrough is eliminated.
Luke Chimuka, Ewa Cukrowska, and Hlanganani Tutu are with the School of Chemistry at the University of Witwatersrand, Johannesburg-South Africa.
Titus A. M. Msagati is with the School of Chemistry, University of KwaZulu-Natal, Durban, Republic of South Africa.
Ronald E. Majors
"Sample Prep Perspectives" Editor Ronald E. Majors is business development manager, Consumables and Accessories Business Unit, Agilent Technologies, Wilmington, DE, and is a member of LCGC's editorial advisory board. Direct correspondence about this column to "Sample Prep Perspectives," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgcmag.com
References
(1) W. Wardencki, J. Curylo, and J, Namiesnik, J. Biochem and Biophys. Methods 70, 275–288 (2007).
(2) S. Mitra, Sample Preparation Methods in Analytical Chemistry (Wiley-Interscience, Hoboken, New Jersey, 2003).
(3) J. Pawliszyn, Sampling and Sample Preparation for Field and Laboratory: Fundamentals and New Directions in Sample Preparation (Elsevier, Amsterdam, Netherlands, 2002).
(4) S. McGillivray, Filtr. & Sep. 43, 36–38 (2006).
(5) A R. Ghiasvand and E. Mohagheghzadeh, Anal. Sci. 20, 917–919 (2004).
(6) C.W. Huie, Anal. and Bioanal. Chem. 373, 23–30 (2002).
(7) R. Krska, J. Chromatogr., A 815, 49–57 (1998).
(8) C. Deng, N. Liu, M. Gao, and X. Zhang, J. Chromatogr., A. 1153: 90–96 (2007).
(9) H. Kataoka, TrAC Trends in Anal. Chem. 22, 232–244 (2003).
(10) P. Chiap, P. Hubert, and J. Crommen, J. Chromatogr., A 948, 151–161 (2002).
(11) H.B. Al-Saffar, B. Ozturk, and R. Hughes, Chem. Eng. Res. and Design 75, 685–692 (1997).
(12) M.Khayet, M.P. Godino, and J.I. Mengual, J. Non-Eq. Thermod. 26, 1–14 (2001).
(13) M. Khayet, A. Velázquez, and J.I. Mengual, J. Non-Eq. Thermod. 29, 279–299 (2001).
(14) L.N. Moskvin and T. G. Nikitina, J. Anal. Chem. 59, 2–16 (2004).
(15) L. Zhu, K. Huey, K.H. Ee, L. Zhao, and H.K. Lee, J. Chromatogr., A 963, 335–343 (2002).
(16) T. Barri and J.å. Jönsson, J. Chromatogr., A 1186, 16–38 (2008).
(17) J.å. Jönsson and L. Mathiasson, J. Chromatogr., A 902, 205–225 (2000).
(18) J.å. Jönsson and L. Mathiasson, in: P.R. Brown and E. Grushka, Eds., Advances in Chromatography (Marcel Dekker, New York, 2001), Chapter 2.
(19) A.A. Ghoreyshi, F.A. Farhadpour, M. Soltanieh, and A. Bansal, J. Membr. Sci. 211, 193–214 (2003).
(20) A.A.Ghoreyshi, F.A. Farhadpour, M. Soltanieh, and M. Abdelghani, J. Membr. Sci. 211, 215–234 (2003).
(21) A. Idris, K.Y. Lee, and H.K. Hing, J. Teknol. 42(F), 35–46 (2005).
(22) K. Johansen, M. Krogh, and K.E. Rasmussen, J. Chromatogr., B, Biomed. Sci. & Appl. 690, 223–231 (1997).
(23) B.Strandberg, P-A. Bergqvist, and C. Rappe, Anal. Chem. 70, 526–533 (1998).
(24) E. Cohen, R.J. Maxwell, and D.J. Donoghue, J. Chromatogr., B. Biomed. Sci. and Appl. 724, 137–145 (1999).
(25) D. Moscone, R.A. Bernardo, E. Marconi, A. Amine, and G. Palleschi, Analyst. 124, 325–329 (1999).
(26) F. Palmisano, M. Quinto, R. Rizzi, and P.G. Zambonin, Analyst 126, 866–870 (2001).
(27) J.å. Jönsson and L. Mathiasson, Trends Anal. Chem. 11, 106–114 (1992).
(28) L. Chimuka, N. Megersa, J. Norberg, L. Mathiasson, and J.å. Jönsson, Anal. Chem. 70, 3906–3911 (1998).
(29) J.å. Jönsson and L. Mathiasson, Trends in Anal. Chem. 18, 318–325 (1999).
(30) L. Chimuka, M.M. Nindi, M.E.M. ElNour, H. Frank, and C. Velasco, J. High Resol. Chromatogr. 22, 417–420 (1999).
(31) J.å. Jönsson and L. Mathiasson, Chromatographia. 52, Supplement 1. S8–S11 (2000b).
(32) L. Chimuka, E. Cukrowska, and J.å. Jönsson, Pure and Appl. Chem. 76, 707–722 (2004).
(33) L. Chimuka, LCGC 22(1), 102–109 (2004).
(34) M. Matsumoto, M..Mikami, and K. Kondo, J. Jap. Petrol. Inst. 49, 256–261 (2006).
(35) J. Norberg and E. Thordarson, Analyst 125, 673–676 (2000).
(36) G. Shen and H.K. Lee, Anal. Chem. 74, 648–654 (2002).
(37) J.X. Wang, D.Q. Jiang, and X.P. Yan, Talanta 68, 945–950 (2006).
(38) E. Miyako, T. Maruyama, N. Kamiya, and M. Goto, Chem. Commun. 7, 2926–2927 (2003).
(39) A. Maximini, H. Chmiel, H. Holdik, and N.W. Maier, J. Membr. Sci. 276, 221–231 (2006).
(40) R. Romero-Gonzalez, E. Pastor-Montoro, J.L. Martinez-Vidal, and A.Garrido-Frenich, Rapid Comm. Mass Spec. 20, 2701–2708 (2006).
(41) A.R. Carabias-Martínez, Rodríguez—Gonzalo, and J. Hernández-Méndez, Anal Chim. Acta 304, 323–332 (1995).
(42) R.G. Melcher, D.W. Bakke, and G.H. Hughes, Anal. Chem. 64, 2258–2262 (1992).
(43) L. Chimuka, T. Nemutandani, E. Cukrowska, and T. Hlanganani, J. Environ. Monitoring 10, 129–135 (2008).
(44) M. Luque, E. Luque-Pérez, A. Ríos, and M. Valcárcel, Anal. Chim. Acta 410, 127–134. (2000).
(45) M. Zougagh, A. Ríos, and M. Valcárcel, Anal. Chim. Acta 539, 117–124 (2005).
(46) T.A.M. Msagati and M.M. Nindi, J. Sep. Sci. 24, 606–661 (2001).
(47) T.A.M. Msagati, and M.M. Nindi, Talanta 69, 243–250 (2006).
(48) T.A.M. Msagati and M.M. Nindi, Talanta, 64, 87–100 (2004).
(49) T.A.M. Msagati and M.M. Nindi, The Bull. Chem. Soc. Japan 78, 2135–2141 (2005).
(50) T.A.M. Msagati and M.M. Nindi, Food Chem. 100, 836–844 (2007).
(51) T.A.M. Msagati and M.M. Nindi, Microchim. Acta 148, 199–214 (2004).
(52) T.A.M. Msagati and M.M. Nindi, Ann. Chim. 96, 635–644 (2006b).
(53) T.A.M. Msagati and M.M. Nindi, Afr. J. Biotech. 5, 1827–1835 (2006).
(54) E. Miyako, T. Maruyama, N. Kamiya, and M. Goto, J. Am. Chem. Soc. 126, 8622–8623 (2004).
(55) R. Romero, J.å. Jönsson, D. Gazquez, M.G. Bagur, and M. Sanchez-vinas, J. Sep. Sci. 25, 584–592 (2002).
(56) V. Jung, L. Chimuka, J.å. Jönsson, N. Niedack, P. Bowens, and B. Alsanius, Anal. Chim. Acta 474, 49–57 (2002).
(57) S. Soko, L. Chimuka, E. Cukrowska, and S. Pole, Anal. Chim. Acta 485, 25–53 (2003).
(58) E.P.S. Ceniceros, A. Ilyina, J.C.C. Esquivel, D.R. Menchaca, J.C.E. Espinoza, and O.E.M. Rodriguez, Vestnik Moscow Univ. Magazine of Ind. Chem., Part 2, 44. Issue 1 (2003).
(59) T.G., Sanchez, J.L.P. Pavon, B.M. Cordero, J. Chromatogr., A 766, 61–69 (1997).
(60) M.M.D. Zamarreño, A.S. Pérez, M.B. Rangel, and J.H. Méndez, Anal. Chim. Acta 386, 99–106 (1999).
(61) M.M.D. Zamarreño, A.S. Pérez, M.B. Rangel, and J.H. Méndez, J. Chromatogr., A 881, 229–241 (2000).
(62) R. Carabias-Martínez, E. Rodríguez—Gonzalo, P.H. Paniagua-Marcos, and J. Hernández-Méndez, J. Chromatogr., A 869, 427–439 (2000).
(63) T. Hyötyläinen, T. Tuutijärvi, K. Kuosmanen, and M.L. Riekkola, Anal. Bioanal. Chem. 372, 732–736 (2002).
(64) D.A. Lambropoulou and T.A. Albanis, J. Biochem. Biophys. Methods 70, 195–228 (2007).
(65) T.G. Bjorhovde, K.E.H. Rasmussen, and S. Pedersen-Bjergaard, Anal. Chim. Acta 491, 155–161 (2003).










New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.


Analytical Challenges in Measuring Migration from Food Contact Materials
November 2nd 2015Food contact materials contain low molecular weight additives and processing aids which can migrate into foods leading to trace levels of contamination. Food safety is ensured through regulations, comprising compositional controls and migration limits, which present a significant analytical challenge to the food industry to ensure compliance and demonstrate due diligence. Of the various analytical approaches, LC-MS/MS has proved to be an essential tool in monitoring migration of target compounds into foods, and more sophisticated approaches such as LC-high resolution MS (Orbitrap) are being increasingly used for untargeted analysis to monitor non-intentionally added substances. This podcast will provide an overview to this area, illustrated with various applications showing current approaches being employed.




Study Explores Thin-Film Extraction of Biogenic Amines via HPLC-MS/MS
March 27th 2025Scientists from Tabriz University and the University of Tabriz explored cellulose acetate-UiO-66-COOH as an affordable coating sorbent for thin film extraction of biogenic amines from cheese and alcohol-free beverages using HPLC-MS/MS.

