Perspectives and Characterization on Antibody–Drug Conjugates
LCGC North America


Comprehensive characterization of ADCs requires increasingly powerful approaches consisting of small- and large-molecule techniques.
This installment of “Perspectives in Modern HPLC” provides an overview of antibody–drug conjugates (ADCs) as a new class of biotherapeutics and describes their analytical characterization for quality assessment with examples from extensive applications libraries.
Chemotherapeutic agents have been the mainstay of anticancer therapy since the early 1940s. Chemotherapy, or the use of cytotoxic agents in medical oncology to inhibit the process of mitotic cell division, is routinely administered with curative intent, to prolong life or as part of palliative care. Although the use of chemotherapy can result in a significant response-for example, in the treatment of testicular cancer-its use is associated with a range of adverse effects. Many of the adverse effects of chemotherapy are the result of damage to healthy cells that divide rapidly and are thus sensitive to antimitotic drugs.
Antibody–drug conjugates (ADCs) are an increasingly important class of biotherapeutics that utilize the specificity of monoclonal antibodies (mAbs) and the cytotoxicity of a potent anticancer payload (1–3). The two molecules are connected via chemical linkers, and the result is a therapy that is able to provide sensitive discrimination between healthy and diseased tissues. The antibody targets and binds to a selected antigenic cell-surface receptor that is, ideally, only expressed on the target cancer cell. After an ADC binds to its target cell, the cell internalizes the ADC through receptor-mediated endocytosis, and the cytotoxic payload is then released inside the lysosomal cellular compartment to provide precise, selective delivery to the cancerous cells. Payload conjugation typically takes place on the amino groups of lysine residues or the sulfhydryl groups of interchain cysteine residues as is the case in ado-trastuzumab emtansine (Kadcyla, Genentech/Roche) and brentuximab vedotin (Adcetris, Seattle Genetics/Millennium Pharmaceuticals), respectively. With 80–100 lysine residues and only eight interchain cysteine residues available in each mAb molecule, lysine conjugation yields a more heterogeneous mixture of species compared to cysteine-conjugated ADCs. Figure 1 depicts examples of common payload conjugation types, namely lysine, cysteine, and glycoconjugates (4).


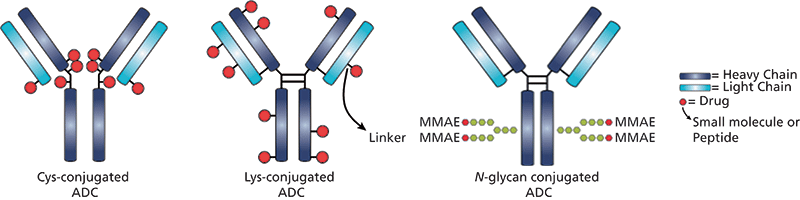
Figure 1: ADC structures showing different sites of attachment to mAb of the drug with the linker. MMAE = monomethyl auristatin E, an extremely potent synthetic antineoplastic agent.
In addition to the described primary amino acid structure, mAbs and ADCs also have distinct higher order structures that dictate their function and immunogenicity. They may be influenced by the above-described modifications and can appear as dimers or aggregates that also have the potential to induce immune responses and affect clearance rates.
For an ADC to demonstrate efficacy, it must incorporate a mAb that recognizes a specific tumor-associated antigen, a linker that has systemic stability but is specifically released at the target cell, and a cytotoxic agent that exhibits toxicity to the tumor cell as a stand-alone modality.
ADC Regulations
Whether submitting to the United States Food and Drug Administration (U.S. FDA), European Medicines Agency (EMA), or other regulatory bodies, ADC developers are covering new territory. Since ADCs incorporate both biologics and small-molecule moieties, these complex therapeutics are difficult to characterize, and multiple health authority experts are required to evaluate different aspects of the end product.
An ADC may be based on a previously approved mAb. For example, trastuzumab (Herceptin) is the mAb portion of the ADC Kadcyla. In such instances, new analytical technologies that have emerged since the development of the original mAb drug product should be evaluated for use in characterizing the related ADC. Consistent with the principles of quality by design (QbD), regulators expect sponsors to use the most current and effective technologies available to build product and process knowledge into controlling product quality.
With the approvals of Kadcyla, Adcetris, and more recently inotuzumab ozogamicin (Besponsa, Pfizer), gemtuzumab ozogamicin (Mylotarg, Pfizer), and more than 50 ADCs in clinical trial pipelines, the clinical application of ADCs is accelerating rapidly (5).
It is important to have a clear understanding of the relationship between the conjugation and manufacturing process, and the resulting product quality and heterogeneity of the ADC. The potency of an ADC is due, in part, to the extent of drug-linker incorporation on the mAb. Methods that can structurally characterize the drug load and distribution have been developed and proven to be critically important for understanding ADC product quality. Wakankar and colleagues have summarized several considerations for the development of analytical methods that measure quality attributes unique to ADCs, such as drug load and drug distribution (6). In addition, several articles documenting the analytical strategies (7) as well as chromatographic and electrophoretic techniques for the characterization of ADCs have been published (8–10).
Characterization and Quality Control Requirements
Quality control (QC) testing of an ADC needs to account for its identity, purity, concentration, and activity (potency or strength)-the same as for any other biopharmaceutical product. Because of the inherent structural complexity of mAbs along with the covalently linked cytotoxic agents, several QC tests are required (8–10). A full understanding of the manufacturing process and its effect on the physicochemical and biological attributes of an ADC must be ascertained. However, in the case of ADCs, even the well-established QC terminology is not straightforward-for instance, the terms potency and strength have different meanings depending on whether the molecule being developed is large or small. The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Q6A for small molecules lists strength (or assay) as a measure of the amount of an active pharmaceutical ingredient (API) (11). ICH Q6B for large molecules uses the term potency as a quantitative measure of biological activity (12). For an ADC that includes both of these components, total function (or potency) would need to be measured with a cell-based assay that assesses overall structure, antigen binding, drug loading, and drug delivery.
Unlike their pharmaceutical predecessors and more straightforward protein-based therapeutics, there is limited availability of certified standards for ADC test method development or comparison. Recently, Merck launched SigmaMAb Antibody-Drug Conjugate Mimic for use as a standard for mass spectrometry (MS) and high performance liquid chromatography (HPLC). SigmaMAb is an "ADC mimic" that conjugates SigmaMab (MSQC4), an IgG1 mAb, to dansylcadaverine fluorophores via a succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) crosslinker (13). At this time, the onus is completely on the developers to devise and implement a set of critical tests for identity and purity, involving the most appropriate analytical technologies. Each intermediate (mAb, linker, and drug) should have a reference standard in addition to an ADC reference standard, to be used in designated release and stability tests. These standards are critical reagents used for analytical method system suitability and in characterization, stability, and bridging studies, as is currently expected for all pharmaceutical and biopharmaceutical products. The cohort of tests would be performed as part of chemistry, manufacturing, and control (CMC) efforts during drug development. Many of these tests would become assays for critical quality attributes (CQA) or analytical methods for specification testing in lot release.
Small-molecule conjugation to mAbs, using any type of strategy, has enormous potential to produce several variant isoforms. Appropriate tests are needed to measure heterogeneity and ensure product consistency. Routine QC testing and characterization may measure the following characteristics:
- Aggregates and fragments
- Charge variants
- Free drug
- Average drug-to-antibody ratio (DAR)
- Drug load distribution, including unconjugated mAb
- Endotoxins or bioburden
Because of the heterogeneity of ADCs, isoforms derived from mAb glycosylation and other post-translational modifications (PTMs) are often controlled at the point of mAb release. The inclusion in the certificate of analysis (CoA) for routine testing of other product-related impurities-such as aggregates, fragments, charge variants, and unconjugated antibodies-discussed above should be assessed product by product. For example, data could be generated to show that an unconjugated antibody is adequately monitored and controlled as part of DAR testing.
Chemical impurities other than free drug or drug-related substances may be evaluated with both ICH Q3B (R2) limits and pharmacology or toxicology input for the specific product (14). Some process-related impurities might be omitted from release testing with sufficient data and process experience over multiple ADC lots or multiple ADC products using the same conjugation platform.
Regulators consider compendial monographs, which exist for small-molecule intermediates, to be the minimum standard for chemical components when used in ADCs.
Drug and Linker: Approaches and Chemistries
The conjugation of anticancer payloads to lysine or cysteine residues found in mAbs results in the generation of ADCs that exhibit significant heterogeneity, with some of the ADC potentially having altered antigen-binding properties leading to suboptimal potency, solubility, stability, and pharmacokinetics. To reduce heterogeneity, expand payload options, and prolong circulating stability, novel site-specific conjugation approaches are actively being pursued within the field (15).
The hydrophobic nature of the payloads used in current ADCs leads to the creation of conjugates of increasing hydrophobicity versus their starting mAb scaffolds. The hydrophobicity of ADCs can promote aggregation, which in turn can lead to hepatotoxicity (16) or increased immunogenicity (17). The hydrophobicity of ADCs can also promote drug resistance via increased affinity for multidrug resistance transports, with the incorporation of hydrophilic linker chemistries shown to bypass multidrug resistance (18).
ADCs use three main tumor-specific microenvironmental factors to selectively release their cytotoxic payloads: cleavable linkers exhibiting protease-sensitivity, pH-sensitivity, and glutathione-sensitivity. Within each of these main linker release mechanisms, significant linker technology advancements are ongoing.
Among the types of conjugation chemistries, enzyme-based site-specific modification shows great potential by eliminating the potential interruption of an antibody–antigen interaction and providing a highly reproducible and modular conjugation system when compared to standard lysine and cysteine conjugation.
Developments in linker chemistries also provide a greater opportunity to incorporate increasingly potent cytotoxic payloads. Quaternary ammonium linkers now enable stable conjugation of payloads with tertiary amine residues (19); the extremely potent synthetic antineoplastic agent monomethyl auristatin E (MMAE) has been linked to mAbs via a linker that is selectively cleaved by cathepsin (for example, in Adcetris) upon entrance into the tumor cell (20). A conjugate with the potent maytansinoid DM1 has been approved (for example, Kadcyla), and Seattle Genetics recently published work on a novel methylene alkoxy carbamate (MAC) self-immolative unit for hydroxyl-containing payloads within ADCs (21). The latter compound enables direct conjugation of drugs through alcohol functional groups that are present on a diverse range of synthetic drugs as well as natural cytotoxic products. Most recently, Spirogen (now part of the AstraZeneca Group) developed a potent and flexible class of ADC payload based on a proprietary pyrrolobenzodiazepine (PBD) technology. PBDs are a family of sequence-selective DNA minor-groove binding agents and are among the most cytotoxic agents known. They are ideally suited for antibody–drug conjugation because of their unique mechanism of action that retains activity against cancer stem cells and is compatible with multiple linker and conjugation technologies. There are two ADCs currently undergoing clinical trial from the collaborative efforts of Spirogen and Seattle Genetics (22), and many more are in the pipeline. As previously mentioned, most of the payload and linker technologies used or studied today impart increasing levels of hydrophobicity on the mAb scaffold (10); for example, DM1 has an estimated LogP value of 3.95 per molecule incorporated. PBDs are even more hydrophobic, with an estimated LogP value of 5.08 per incorporated molecule. To address this issue, hydrophilic spacers (for example, para-aminobenzyl alcohol [PAB]) and linkers (such as polyethylene glycol [PEG]) are often incorporated as part of the bioconjugation chemistry to balance out the increased hydrophobicity introduced by the conjugation of the payload.
Chromatography for mAb, Drug, Linker, and ADC
Various ultrahigh-pressure liquid chromatography (UHPLC) techniques have proved to be useful for analyzing ADC heterogeneity at the intact level, including hydrophobic-interaction chromatography (HIC), ion-exchange chromatography (IEC), size-exclusion chromatography (SEC), and reversed-phase chromatography. Where appropriate, the coupling of these separation techniques with high-resolution accurate mass spectrometry (HRAM MS) presents a powerful characterization tool. Further structural details can be ascertained by breaking down the intact ADC; both peptide mapping using reversed-phase chromatography and released glycan analysis with hydrophilic-interaction chromatography (HILIC) are deemed essential tools. Each of these analytical approaches reveals different CQAs of the ADC-from primary amino acid sequence and associated modifications (peptide mapping) to the presence of higher order aggregated structures (SEC) that could impact product efficacy and safety. In addition to the standard cohort of small molecule and large biomolecule characterization methodologies, a whole set of tests must be performed to interrogate the level of drug conjugation and the levels of unconjugated mAb, payload, and linker (as shown in Figure 2).

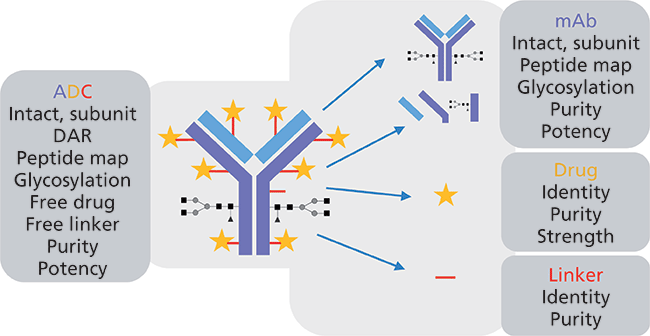
Figure 2: Typical characterization approaches performed on ADC therapeutics.
Monoclonal Antibody Primary Sequence Analysis
As a technique, peptide mapping is well established in the biotechnology industry with roots lying in protein characterization, proteomics, and de novo peptide sequencing. In recent years, advances in sample preparation (protein digestion), peptide separation, HRAM MS capabilities, and bioinformatics have enabled the biotech industry to confidently apply peptide mapping workflows in routine, high-throughput environments.
Peptide mapping can reveal many CQAs of a protein. In the case of ADCs, peptide mapping is fundamental in confirming not only the sequence of the mAb, but also the site and level of drug conjugation (Figure 3). The accuracy with which this information can be determined is based on the method of protein digestion and fidelity of the subsequent UHPLC and MS analysis. The type of fragmentation used within the MS system should also be carefully considered because standard collision-induced dissociation (CID) experiments often fail to reveal the precise site of drug conjugation or glycosylation. Alternative or additive fragmentation techniques such as higher energy collisional dissociation (HCD), electron transfer dissociation (ETD), and ultraviolet photodissociation (UVPD) are becoming increasingly important in the elucidation of site-specific modifications and can generate informative fragmentation patterns, even at the subunit level (23–25).

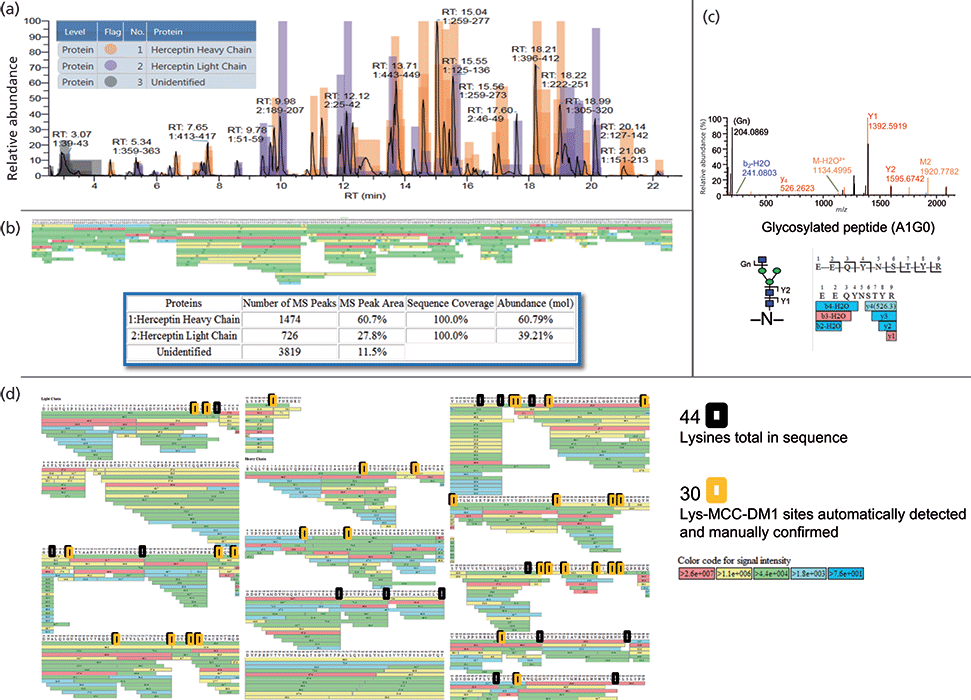
Figure 3: Trastuzumab emtansine lysine-conjugation mapping (26). (a) Color-coded base peak ion chromatogram (BPI) showing heavy and light chain peptides. (b) Coverage map showing 100% sequence coverage, number of MS peaks, and relative abundance of heavy and light chain peptides detected. (c) Example higher energy collisional dissociation (HCD) MS/MS spectrum of a glycopeptide showing fragmentation of both peptide and glycan. (d) Identification of lysine conjugated MCC-DM1 at the peptide level.
Chromatographic Techniques for the Determination of Antibody Variants, Fragments, DAR, and Payload Mapping
Hydrophobic Interaction Chromatography
HIC separates proteins by the interactions between hydrophobic pockets present on the surface of the protein and the hydrophobic ligands on the HIC resin. Proteins are loaded onto the column in relatively high salt concentrations to induce hydrophobic interactions and are eluted by reducing the salt concentration of the mobile phase during the chromatographic separation. The binding of the proteins is dependent on the inherent surface hydrophobicity, which is influenced by the conformation of the protein. Changes in protein conformation can be characterized by this mode of chromatography, and several publications exist that indicate that common modifications of mAbs, such as oxidation and deamidation, can be seen using HIC (27). With the conjugation of hydrophobic payloads to the mAb to form ADCs, the use of HIC for DAR analysis has become increasingly popular (6,28).

With each additional linkage of the drug to the mAb the retention of the ADC species on the column increases, thus allowing quantification of drug load on the ADC and resolution of isomeric configurations of the same DAR (Figure 4).

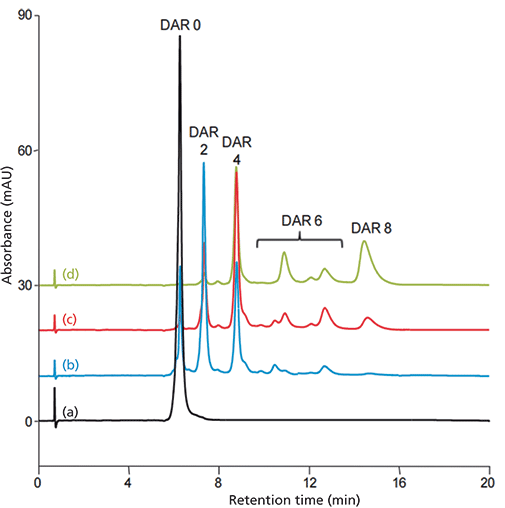
Figure 4: Comparison of synthesized Cys-conjugated ADC mimics with different drug load (29): (a) unconjugated mAb (5 mg/mL), (b) Cys-conjugated ADC mimic (low load, 5 mg/mL), (c) Cys-conjugated ADC mimic (moderate load, 5 mg/mL), (d) Cys-conjugated ADC mimic (high load, 5 mg/mL). Column: 100 mm × 4.6 mm, 5-µm dp Thermo Scientific MAbPac HIC-Butyl; mobile-phase A: 95:5 (v/v) 1.5 M ammonium phosphate (pH 7.0)–isopropanol; mobile-phase B: 80:20 (v/v) 50 mM sodium phosphate (pH 7.0)–isopropanol; gradient: 0% B for 6 min, 0–100% B in 14 min, hold at 100% B for 5 min; temperature: 25 °C; flow rate: 1.0 mL/min; injection volume: 5 µL (5 mg/mL); detection: UV absorbance at 280 nm.
Ion-Exchange Chromatography
IEC involving cation-exchange column chemistries is a standard method used to separate and monitor the charge-variant profile of mAb-based therapeutics (30). Charge-variant separations have been further developed with the use of pH gradients that provide ease of use and a more global approach to the method development process (Figure 5) (31). There are several PTMs that can alter the charge or conformation of a protein and can, therefore, be characterized using IEC. Glycan variants, deamidation, oxidation, and even aggregation are among them. The specific charge-variant profile that is obtained from a mAb is closely monitored at each stage in the production to ensure the product quality remains the same. In the case of ADCs, mAbs may not provide an informative charge-variant profile-if the drug or linker is charged, or linkage occurs through a charged amino acid (such as lysine), the underlying mAb charge heterogeneity is difficult to assess because conjugation affects the overall charge of the conjugated molecule. In such cases, the "charge profile" is often more of a "conjugation profile." Despite this, measuring the distribution of charged species can be a good way to demonstrate process consistency and thus should be included in an ADC comparability toolkit.

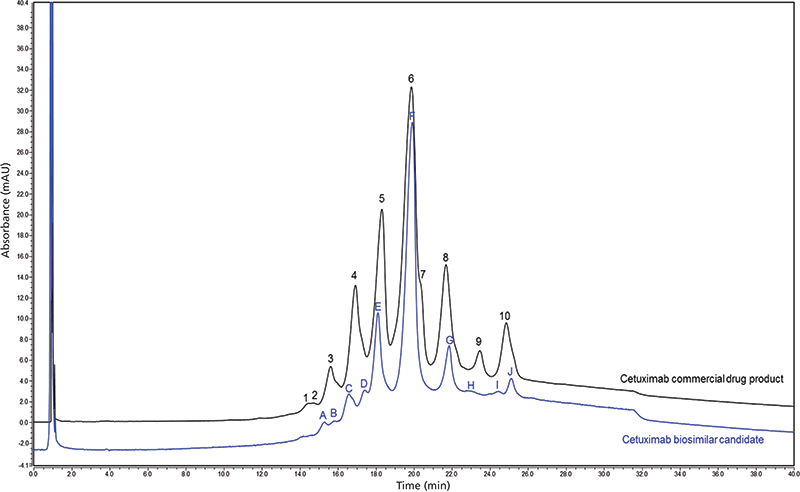
Figure 5: Charge variant chromatographic profile comparison of commercial chimeric IgG1 mAb (black trace) and cetuximab biosimilar candidate (blue trace) obtained with cation-exchange chromatography in pH-based gradient mode (31). Peak labeling corresponds to the number of peaks in each trace and does not indicate peak identification.
Reversed-Phase Chromatography-MS
MS analysis of ADC drug distribution provides insights into the relative concentration of different drug-linked forms, which may elicit distinct pharmacokinetic and toxicological properties. MS analysis of ADC drug distribution is particularly advantageous for conjugates produced using linkage through surface-accessible lysine residues, which are not easily separated by chromatography alone because of their high degree of heterogeneity.
Reversed-phase LC–MS can be used to elucidate the positional isomers of ADCs. Reversed-phase LC–MS following IdeS proteolytic digestion facilitates the subunit analysis of ADCs and enables rapid comparison of the ADC samples, for instance for batch assessment (Figure 6). Indeed, IdeS proteolytic digestion has been proposed as an analytical reference method at all stages of ADC discovery, preclinical and clinical development, for routine comparability assays, formulation, process scale-up and transfer, and to define CQAs in a QbD approach (32).

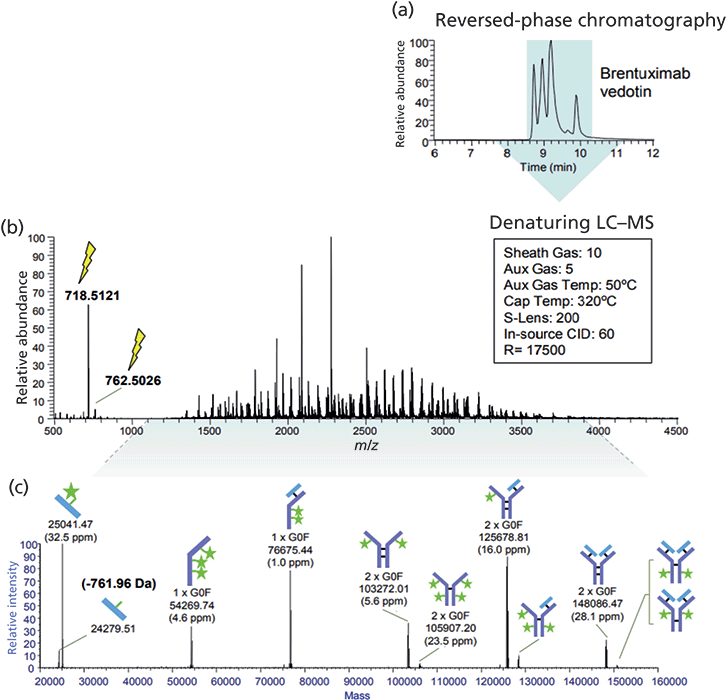
Figure 6: Denaturing LC–MS analysis of the ADC brentuximab vedotin (Adcetris) (33). (a) Unmodified sample (1 µg) was analyzed by reversed-phase chromatography coupled to an orbital trap MS system produced several peaks. (b) The resulting averaged MS spectrum is a complex mixture of charge state envelopes as well as a vcMMAE-specific reporter fragment ion at m/z 718. (c) Data analysis with ReSpect deconvolution and Sliding Window integration show roughly six covalently-structured forms of unraveled cysteine-linked ADC.
Chromatography and Native Mass Spectrometry
The ADCs currently approved for use utilize naturally occurring lysine side chain amino groups or the cysteine thiol groups, which are formed upon partial reduction of IgG intramolecular disulfide bonds, for conjugation of the drug load (34).
Cysteine-linked ADCs present a unique challenge for characterization because proper intact analysis requires native MS conditions to preserve structurally critical noncovalent binding between antibody chains.
ADCs exhibit significant heterogeneity resulting from the number and distribution of drug molecules across the antibody. This level of molecular complexity and heterogeneity presents a considerable challenge for current analytical techniques.
Native MS of intact proteins allows direct observation of molecules that rely on noncovalent interactions to preserve critical structural features, such as interchain associations that hold together cysteine-linked ADCs. The use of 100% aqueous and physiological pH buffers in native MS analysis produces decreased charge states (increased m/z) and improves mass separation of heterogeneous mixtures.
An orbital trap native MS workflow has recently been developed that is compatible with SEC, allowing online desalting and sample delivery, to observe intact proteins at high m/z ranges. This strategy reduces mass interference in complex protein spectra by increasing peak capacity in the m/z space. This workflow has recently been applied to the analysis of Adcetris and Kadcyla, cysteine-linked and lysine-linked ADCs, respectively, and the accurate calculation of DAR (Figure 7).

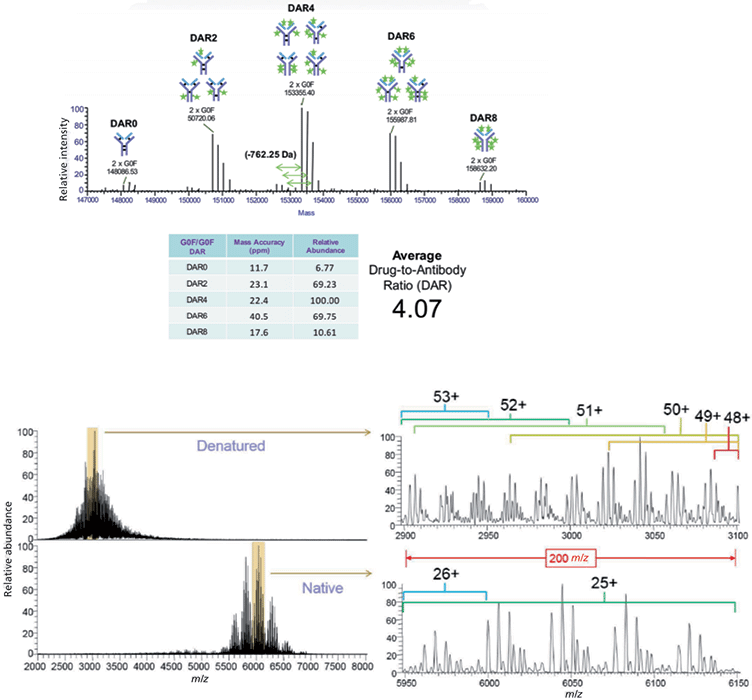
Figure 7: Desalting SEC–MS DAR of Adcetris and Kadcyla (35). (a) Desalting SEC is compatible with a native MS approach and allows preservation of noncovalent interactions which support the structure of cysteine-linked ADCs. Based on the individual deconvoluted abundances of the G0F/G0F glycoform, the authors calculated an average DAR value of 4.07 (32). (b) Denaturing MS spectra (from reversed-phase LC) are observed at lower m/z ranges while native MS spectra from online SEC are observed at higher m/z ranges. A detailed view shows that 2–3 sequential charge state envelopes overlap compared to an overlap of 0– charge state envelopes in the native MS spectrum.
This work built on a similar approach that was first applied to the study of Adcetris using an orbital trap mass spectrometer equipped with a high-mass quadrupole mass selector (36).
Higher Order Structural Analysis
Hydrogen–deuterium exchange (HDX)-MS is a powerful tool for studying the dynamics of higher-order structure of protein-based therapeutics. The rate of hydrogen-to-deuterium exchange within the amide hydrogen on the backbone of biotherapeutics provides solvent accessibility information, and thus protein structure and conformation can be inferred.
Although HDX-MS cannot be used to define an absolute structure in the manner of X-ray crystallography, it can be used to directly assess the native structure in a comparative fashion. Proteins in solution are highly dynamic, and the stability and functionality of any protein therapeutic are closely associated to a specific conformation.
The manufacturing of ADCs involves additional processing steps during conjugation, and it is important to evaluate how the drug conjugation process impacts the conformation and dynamics of the mAb intermediate. The ability of HDX-MS to monitor conformational changes at the peptide level makes the technique well-suited for providing detailed insights into the impact of drug conjugation processes on the higher-order structure of mAbs.
Orbital trap–based HDX-MS has previously been used to probe the conformation and dynamics of interchain cysteine-linked ADCs (37). In this publication, a side-by-side HDX comparison of ADCs, mAbs, reduced mAbs, and partially reduced mAbs was used to identify minor local conformational changes and confirm that these were because of the partial loss of interchain disulfide bonds in ADCs. These findings were used to indicate that ADC manufacturing processes that involve partial reduction of mAb interchain cysteine residues followed by conjugation with drug linkers do not significantly impact the conformational integrity of the mAb. A similar approach has been used to study the antibody structural integrity of site-specific ADCs (38). Together these results highlight the utility of HDX-MS for interrogating the higher-order structure of ADCs and other protein therapeutics.
Residual Free Drug Analysis and Control Strategy for Small-Molecule Impurities in ADCs
On the analytical front, one approach to conducting free-drug analysis for ADC drug substance and drug-product preparations is to precipitate the proteins (along with protein-bound drug) and analyze the resulting supernatant using a method that is effective for detecting the small molecule such as those using UHPLC–MS or UHPLC with ultraviolet (UV) detection.
Residual Solvents and Volatile Organic Impurities in ADCs
It is uncommon that residual solvent analysis is conducted for post-production quality assurance of conventional protein-based biopharmaceuticals such as mAbs. Organic solvents are not typically used in cultured cell trains and seldom form part of the risk profile of the drug.
In contrast, the conjugation reaction to form ADCs generally involves a site-selective enzymatic or chemical reaction of antibody to linker to small-molecule drug warhead, where the hydrophobic warhead and linker are solubilized in solvents such as N,N-dimethylacetamide (DMA), N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), or propylene glycol (PG). The conjugation process is followed by protein purification techniques to remove process-related contaminants (unconjugated toxin and residual solvents). However, strategies must be in place to monitor for such impurities. For the analysis of these residual solvents, one possible approach is to use a direct gas chromatography (GC) technique (43) after removal of the proteins rather than the traditional headspace GC approach in USP <467> (44). Because of the low levels expected for residual solvents in ADC samples, an alternative GC–MS method (particularly using the selected ion monitoring mode) is likely to yield higher sensitivity as well as provide identification information on unknown peaks, as shown in the example in Figure 8.

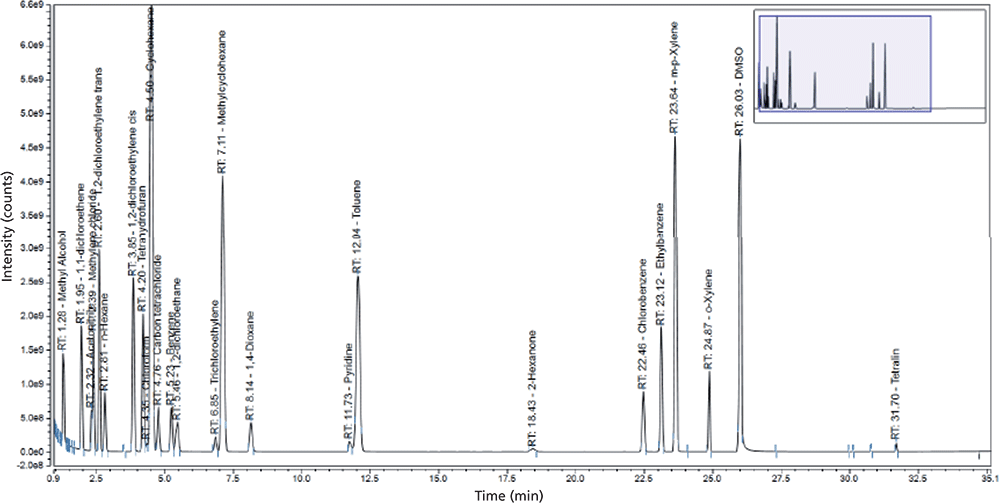
Figure 8: GC–MS of residual solvents following analytical headspace GC conditions similar to those in USP <467> that may provide higher sensitivity under single ion monitoring mode as well as information for unknown peak identification.
Bioanalysis of ADCs
ADCs are complex heterogeneous mixtures resulting from differences in glycosylation of the antibody, the number of small-molecule drug moieties attached to the antibody, and the location of the conjugation sites. This situation is further complicated as the drug undergoes in vivo changes such as spontaneous deconjugation of the small-molecule drug and differential clearance rates of ADC components as a result of their different DARs. These changes, as well as other attributes of ADCs, contribute to the unique challenges in their bioanalysis. Furthermore, it is becoming clearer that the data required by the bioanalytical scientist is also dependent on the phase of the ADC development. The early discovery phase requires in vivo stability of ADC candidates based on monitoring average DAR or presence and integrity of the drug moiety at a specific conjugation site, while in the clinical development phase, it is important to establish a correlative relationship between one or more components of the ADC and various safety and efficacy indicators. Therefore, to address these bioanalytical challenges both ligand binding assays (LBAs) and LC–MS have been used. For instance, measurement of total antibody to assess antibody pharmacokinetic (PK) behavior and measurement of conjugated antibody (DAR ≥ 1) is typically performed using LBAs, with unconjugated drug monitored by LC–MS. However, a hybrid of the two approaches, referred to as hybrid LC–MS, is becoming more actively developed and applied in ADC bioanalysis. This platform uses the affinity capture of the LBA to retain sensitivity and LC–MS for detection to provide greater specificity and improved characterization of the ADC component being monitored. Therefore, the hybrid LC–MS approach provides benefits of both the LBA and LC–MS, enabling scientists to better address some of the unique challenges of ADC bioanalysis and to allow for the use of a single platform to generate the data required for ADC bioanalysis (45).
Summary
ADCs are an increasingly important class of biotherapeutics. As the list of the first-generation ADCs entering the clinic grows, new generations of ADCs will benefit from their insights. The future looks set to see ADCs that have higher levels of cytotoxic drug conjugation, lower levels of unconjugated antibodies, more-stable linkers between the drug and the antibody, and increasing analytical challenges. The stability of linkers in circulation is critical to ensure patient safety and to mitigate the side effects caused by the off-target release of toxic payloads.
Today's ADCs pose unique analytical challenges requiring increasingly powerful approaches, consisting of small- and large-molecule techniques for their comprehensive characterization. The complexity of their analysis is matched only with their potential to become the "magic bullet" of anticancer treatment.
Acknowledgments
The authors thank Davy Guillarme and Szabolcs Fekete of the University of Geneva, Michael Heidorn of Thermo Fisher Scientific, Tao Chen and Chris Yu of Genentech, Francois D'Hooge of Novasep, He Meng of Sanofi, Srikanth Kotapati of BMS, and Yanqun Zhao of AbbVie for providing technical and editorial inputs to the manuscript.
References
(1) Antibody-Drug Conjugates: Fundamentals, Drug Development, and Clinical Outcomes to Target Cancer, K.J. Olivier and S.A. Hurvitz, Eds. (Wiley, Hoboken, New Jersey, 2017), pp. 1–560.
(2) Antibody-Drug Conjugates: The 21st Century Magic Bullets for Cancer, J. Wang, W-C. Shen, and J. L. Zaro, Eds.(AAPS Advances in the Pharmaceutical Sciences Series, Springer, Heidelberg, Germany, 2015).
(3) N. Diamantis and U. Banerji, Br. J. Cancer 114(4), 362–367 (2016).
(4) H.J. Wang, T. Zhang, B.J. Agnew, R. Viner, S. Lin, S. Houel, and J. Josephs, “Orbitrap Based Mass Spectrometric Characterization of Antibody Drug Conjugates Engineered through Antibody Glycans,” Thermo Scientific Poster Note, accessed online January 2018 (https://tools.thermofisher.com/content/sfs/posters/PN-64397-LC-MS-ADC-Antibody-Glycans-AAPS2015-PN64397-EN.pdf).
(5) R.S. Zolot, S. Basu, and R.P. Million, Nat. Rev. Drug Discovery 12, 259–260 (2013).
(6) A. Wakankar, Y. Chen, Y. Gokarn, and F.S. Jacobson, MAbs 3, 161–172 (2011).
(7) A. Beck, L. Goetsch, C. Dumontet, and N. Corvaïa, Nat. Rev. Drug Discovery 16(5), 315–337 (2017).
(8) B. Bobály, S. Fleury-Souverain, A. Beck, J.L. Veuthey, D. Guillarme, and S. Fekete, J. Pharm. Biomed. Anal. 147, 493–505 (2018).
(9) T. Chen, Y. Chen, C. Stella, C.D. Medley, J.A. Gruenhagen, and K. Zhang, J. Chromatogr. B 1032, 39–50 (2016).
(10) R. Neupane and J. Bergquist, Eur. J. Mass Spectrom. 23(6), 417–426 (2017).
(11) International Conference on Harmonization, ICH Harmonised Tripartite Guideline, ICH Q6A Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances (ICH, Geneva, Switzerland, 2000).
(12) International Conference on Harmonization, ICH Harmonised Tripartite Guideline, ICH Q6B: Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products (ICH, Geneva, Switzerland, 1999).
(13) SigmaMAb Antibody-Drug Conjugate (ADC) Mimic product webpage, Sigma-Aldrich, United Kingdom, accessed online January 2018 (https://www.sigmaaldrich.com/catalog/product/sigma/msqc8?lang=en®ion=GB).
(14) International Conference on Harmonization, ICH Harmonised Tripartite Guideline: Impurities in New Drug Products, ICH Q3B (R2) (ICH, Geneva, Switzerland, 2006).
(15) J.R. McCombs and S.C. Owen, AAPS J. 17(2), 339–351 (2015).
(16) L.M. Jarvis, Chem. Eng. News 90(25), 12–18 (2012).
(17) M.K. Joubert et al., J. Biol. Chem. 287(30), 25266–25279 (2012).
(18) Y.V. Kovtun et al., Cancer Res. 70, 2528–2537 (2010).
(19) P.J. Burke, J.Z. Hamilton, T.A. Pires, J.R. Setter, J.H. Hunter, J.H. Cochran, A.B. Waight, and K.A. Gordon, Mol. Cancer Ther. 15(5), 938–945 (2016).
(20) J.A. Francisco et al., Blood. 102(4), 1458–1465 (2003).
(21) R.V. Kolakowski, K.T. Haelsig, K.K. Emmerton, C.I. Leiske, J.B. Miyamoto, J.H. Cochran, R.P. Lyon, P.D. Senter, and S.C. Jeffrey, Angew Chem. Int. Ed. Engl. 55(28), 7948–7951 (2016).
(22) J. Mantaj, P.J.M. Jackson, K.M. Rahman, and D.E. Thurston, Angew Chem. Int. Ed. Engl. 56(2), 462–488 (2017).
(23) J. Qian and M. Sanders, “Degradation Profiling of a Monoclonal Antibody Using Multiple Fragmentation Techniques and a Novel Peptide Mapping Software,” Thermo Fisher Scientific Poster Note, Accessed online October 2017 (https://tools.thermofisher.com/content/sfs/posters/PN-64146-Degradation-Profiling-ASMS2014-PN64146-EN.pdf).
(24) J.S. Brodbelt, Chem. Soc. Rev. 43(8), 2757–2783 (2014).
(25) S. Lu, S.-B. Fan, B. Yang, Y.-X. Li, J.-M. Meng, L. Wu, P. Li, K. Zhang, M.-J. Zhang, Y. Fu, J. Luo, R.-X. Sun, S.-M. He and M.-Q. Dong, Nat. Methods 12, 329–331 (2015).
(26) A.O. Bailey, S. Houel, E. Damoc, K. Scheffler, and J.L. Josephs, “Integrated Characterization of a Lysine-Linked Antibody-Drug Conjugate by Native Intact Mass Analysis and Peptide Mapping Performed on a Hybrid Quadrupole-Orbitrap Mass Spectrometer with High Mass Range,” Thermo Scientific Poster Note, accessed online January 2018. (https://tools.thermofisher.com/content/sfs/posters/PO-72282-LC-MS-Lysine-Linked-ADCs-ATE2017-PO72282-EN.pdf)
(27) J.V. Douglass, A. Wallace, and A. Balland, J. Chromatogr. A 1214(1-2), 81–89 (2008).
(28) A. Cusumano, D. Guillarme, A. Beck, and S. Fekete, J. Pharm. Biomed. Anal. 121, 161–173 (2016).
(29) J. Baek, I. Birznieks, S. Lin, and X. Liu, “Analysis of Monoclonal Antibodies and Antibody-Drug Conjugates Using New Hydrophobic Interaction Chromatography (HIC) Columns,” Thermo Scientific Poster Note, accessed online January 2018 (https://tools.thermofisher.com/content/sfs/posters/PN-21218-HPLC-Monoclonal-Antibodies-HIC-PN21218-EN-Rev1.pdf).
(30) M. Weitzhandler, D. Farnan, J. Horvath, J.S. Rohrer, R.W. Slingsby, N. Avdalovic, and C. Pohl, J. Chromatogr. A 828(1–2), 365–372 (1998).
(31) S. Millán, A. Trappe, A. Farrell, and J. Bones, “Simple Charge Variant Profile Comparison of an Innovator Monoclonal Antibody and a Biosimilar Candidate,” Thermo Scientific Application Note (in press, 2018).
(32) E. Wagner-Rousset, M.-C. Janin-Bussat, O. Colas, M. Excoffier, D. Ayoub, J.-F. Haeuw, I. Rilatt, M. Perez, N. Corvaïa, and A. Beck, MAbs 6(1), 173–184 (2014).
(33) “Cysteine Linked Antibody-Drug Conjugate Performed on a Hybrid Quadrupole-Orbitrap Mass Spectrometer with High Mass Range,” Thermo Scientific Poster Note, accessed online January 2018 (https://assets.thermofisher.com/TFS-Assets/CMD/posters/PN-64802-LC-MS-Cysteine-linked-Antibody-Drug-Conjugate-ASMS2016-PN64802-EN.pdf).
(34) A. Beck, J.-F. Haeuw, T. Wurch, L. Goetsch, C. Bailly, and N. Corvaïa, Discovery Med. 10(53), 329–339 (2010).
(35) A.O. Bailey, S. Houel, K. Scheffler, E. Damoc, J. Sutton, and J. L. Josephs, “Complete Characterization of a Lysine-Linked Antibody-Drug Conjugate by Native LC/MS Intact Mass Analysis and Peptide Mapping,” Thermo Scientific Application Note, accessed online January 2018 (https://assets.thermofisher.com/TFS-Assets/CMD/Application-Notes/an-72511-lc-ms-lysine-linked-adc-an72511-en.pdf).
(36) A. Dyachenko, G. Wang, M. Belov, A. Makarov, R.N. de Jong, E.T. van den Bremer, P.W. Parren, and A.J. Heck, Anal. Chem. 87(12), 6095–6102 (2015).
(37) L.Y. Pan, O. Salas-Solano, and J.F. Valliere-Douglass, Anal. Chem. 86(5), 2657–2664 (2014).
(38) L.Y. Pan, O. Salas-Solano, and J.F. Valliere-Douglass, Anal. Chem. 87(11), 5669–5676 (2015).
(39) H.H. Gong, N. Ihle, M.T. Jones, K. Kelly, L. Kott, T. Raglione, S. Whitlock, Q. Zhang, and J. Zheng, AAPS PharmSciTech 19(3), 971–977 (2018).
(40) International Conference on Harmonization, ICH Harmonised Tripartite Guideline: Impurities in New Drug substances, Q3A (R2) (ICH, Geneva, Switzerland, 2006).
(41) International Conference on Harmonization, ICH Harmonised Tripartite Guideline ICH Q5B: Quality of Biotechnological Products (ICH, Geneva, Switzerland, 1995).
(42) International Conference on Harmonization, ICH Harmonised Guideline ICH M7(R1): Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk (ICH, Geneva, Switzerland, 2017).
(43) C.D. Medley, J. Kay, Y. Li, J. Gruenhagen, P. Yehl, and N.P. Chetwyn, Anal. Chim. Acta 850, 92–96 (2014).
(44) General Chapter <467> “Residual Solvents,” in United States Pharmacopeia 40–National Formulary 35 (United States Pharmacopeial Convention, Rockville, Maryland). Available at:
.
(45) K. Antwi, A. Ribar, U. Kiernan, and E.E. Niederkofler, “Quantitative Analysis of an Antibody Drug Conjugate using MSIA D.A.R.T.‘S Technology, an Integral Part of the Universal Ligand Binding Mass Spectrometric Immunoassay (LB-MSIA) Workflow,” Thermo Scientific Application Note, accessed online January 2018 (https://assets.thermofisher.com/TFS-Assets/LCD/Application-Notes/LCD-MSIA-AnitibodyDrugConjugate-AppNote-APAACMSIAADCQuan-EN.pdf).
ABOUT THE AUTHORS

Rowan E. Moore

Rowan E. Moore PhD, MRSC, has more than 15 years of experience with LC and high resolution MS and she currently manages a Bio/Pharma applications development team at Thermo Fisher Scientific. She gained her PhD from the University of Liverpool, where she applied proteomic approaches to the identification and detection of pyrrolizidine alkaloidosis biomarkers in equines. Rowan has since worked in regulated bioanalysis and in roles for leading instrument manufacturers, maintaining a keen focus on the delivery of analytical solutions for customer success.
Kelly Broster
Kelly Broster PhD, is a former Cancer Research UK scientist, now focused on biopharmaceutical and pharmaceutical applications with a focus on customer needs in LC and MS at Thermo Fisher Scientific. Kelly has an MSc in Biomedical Science, a PhD in Cancer Proteomics, and held a post-doctoral position in Systems Oncology.
Ken Cook
Ken Cook PhD, has more than 30 years of experience supporting LC and IC applications. Ken currently supports customers in bio-separations for Thermo Fisher Scientific. Previously, Ken was a lecturer in Biochemistry at the University of Newcastle-Upon-Tyne, UK where he focused on protein biochemistry.
Kyle D'Silva
Kyle D'Silva PhD, MRSC, has more than 15 years of experience in GC and HRMS, working for several instrument manufacturers. Kyle is responsible for marketing connected analytical workflow solutions for the pharmaceutical and biopharmaceutical markets at Thermo Fisher Scientific. He holds an MSc in Analytical Chemistry and Instrumentation, and a PhD in research involving applied analytical chemistry and environmental sciences.

Eric Niederkofler
Eric Niederkofler PhD, is the R&D and Applications Manager at Thermo Fisher Scientific, currently managing activities related to the Mass Spectrometric Immunoassay (MSIA) product line. Eric has a PhD in bio-analytical chemistry from Arizona State University, and has authored more than 30 peer reviewed scientific articles and book chapters on his work in the field of targeted proteomics.

Aaron O. Bailey

Aaron O. Bailey PhD, trained in MS in the laboratory of Professor John R. Yates III at The Scripps Research Institute. He earned his doctorate in cell biology at the University of Virginia, then joined Thermo Fisher Scientific as an Applications Scientist focused on large biomolecule analysis. Aaron later joined the biopharma marketing team to lead efforts in intact protein analysis, with particular focus on native MS applications.

Jonathan Bones

Jonathan Bones PhD, is a Principal Investigator at the National Institute of Bioprocessing Research and Training (NIBRT). Jonathan received a PhD in Analytical Chemistry, then undertook postdoctoral research with Professor Pauline Rudd at NIBRT. He then moved to Northeastern University in Boston, working with Professor Barry L. Karger. Jonathan returned to NIBRT in 2012 following receipt of funding from Science Foundation Ireland to form his own independent research group focused on the development and application of advanced LC–MS and CE-MS solutions for the characterization and comparability assessment of therapeutic proteins and manufacturing processes.
ABOUT THE COLUMN EDITOR
Michael W. Dong
Michael W. Dong is a principal of MWD Consulting, which provides training and consulting services in HPLC and UHPLC, method improvements, pharmaceutical analysis, and drug quality. He was formerly a Senior Scientist at Genentech, Research Fellow at Purdue Pharma, and Senior Staff Scientist at Applied Biosystems/PerkinElmer. He holds a PhD in Analytical Chemistry from City University of New York. He has more than 100 publications and a best-selling book in chromatography. He is an editorial advisory board member of LCGC North America and the Chinese American Chromatography Association. Direct correspondence to: LCGCedit@ubm.com

















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.



