Analytical Characterization of Biotherapeutic Products, Part I: Quality Attributes
LCGC North America


To ensure the reliable and accurate characterization of biotherapeutics, an arsenal of orthogonal analytical techniques is needed.
The potential for the presence of multiple impurities and variants in biotherapeutic products highlights the need for having an arsenal of orthogonal analytical techniques that together can yield a reliable and accurate characterization of the product. Here, in the first part of this two-part series, we discuss the various quality attributes that are pertinent to a biotherapeutic. In part II, we will present the current and evolving practices that are being used for analysis of these attributes.
Since the commercialization of human insulin in 1982, the first genetically engineered therapeutic, there has been exponential progress in the field of biotherapeutics, both in terms of the complexity of the molecules produced as well as their characterization. Moreover, as the innovator drugs reach their patent cliff, they have paved the way for biosimilars, which are copies of innovator drugs that offer similar physical and chemical characteristics. However, unlike a "generic" drug that is a chemically synthesized identical copy of the innovator drug, the production of biosimilars is complex because it involves the recombinant expression of the specific protein or peptide in a biological host (1,2). Therefore, the amount and type of permissible structural and functional variations have to be strictly controlled to match the innovator molecule. This requires extensive physicochemical characterization of the structure, structural variation, and impurities that may arise from the manufacturing process itself or the host used for the protein production (3). Typically, these variations can be categorized into those that result in substantial changes to the safety and efficacy profile of the product (product impurities) and those that result in less than substantial changes (product variants) (4).
The potential for the presence of multiple impurities and variants in biotherapeutic products highlights the need for having an arsenal of orthogonal analytical techniques that together can yield a reliable and accurate characterization of the product. Here, we discuss the various quality attributes that are pertinent to a biotherapeutic. In part II of this series, we will present the current and evolving practices that are being used for the analysis of these attributes.
Biotherapeutics as Proteins
Before delving deeper into the quality attributes and their effect on product efficacy and safety, it is important to appreciate that biotherapeutics are essentially proteins or protein complexes arising from a three-dimensional (3D) arrangement of single or multiple linear polypeptide chains, respectively.
Parallels can be drawn between the structural hierarchy of a protein and other forms of coded information. An example of a collection of books can be used to understand this layering of information, where a sentence, although meaningful as a stand-alone, only imparts limited information, and can be used in conjunction with other sentences to create a paragraph that conveys more meaning and context. Similarly, a chapter consisting of many paragraphs would hold more information, and so on (Figure 1). The different levels of structural complexity of a protein are discussed in further detail below and illustrated (Figure 1) using the example of a monoclonal antibody (mAb).


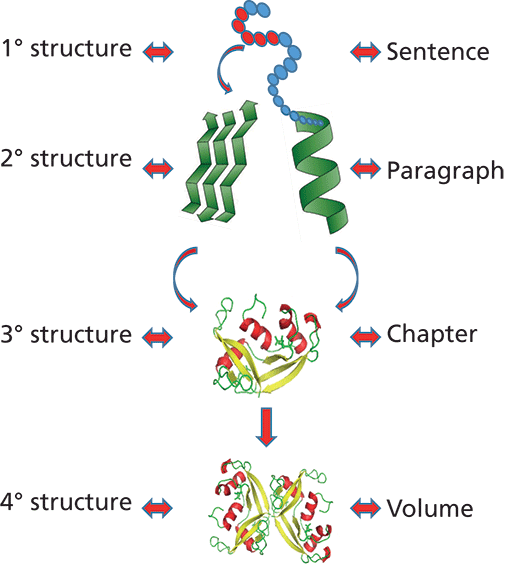
Figure 1: Structural complexity of a mAb. The primary structure consists of a linear chain of amino acids that arrange in three dimensions in a specific manner to form the secondary structure consisting of alpha helix, beta sheets, and random coil. The tertiary structure consists of functionally active arrangements of secondary structures. In mAbs, interactions between a number of the same or different tertiary structures can occur to form the quaternary structure.
Primary Structure
The unique, linear amino acids sequence of a polypeptide, held together by covalent peptide bonds, is known as its primary structure. This sequence is a fingerprint of the protein and defines its structure, function, and, in certain cases, even its intracellular localization. Because different amino acids differ structurally by different side-chain substituents, their unique arrangement confers different structural and functional properties to proteins. The primary structure has two ends, one with a free carboxyl group called the carboxy-terminus or C-terminus, the other with a free α-amino group known as the amino-terminus or N-terminus (5).
In the context of biotherapeutics, the primary structure holds important information like the conserved functional domains (the constant region in mAbs) or the variable region on which the antigen binding sites unique to each antibody are located. The location of conserved cysteine participating in disulfide bond formation (insulin, mAbs) are all identifiable from the primary structure of a biotherapeutic (6). However, it is important to remember that the sequence specificity of this translated linear information is but a prerequisite to a protein and correct processing of higher orders of structure needs to take place for it to attain proper functionality.
Higher Order Structure
Protein higher order structure (HOS) includes all orders of the structure above the primary structure, such as secondary, tertiary, and, in some cases, quaternary structure. HOS assessment is a key component in defining a biotherapeutic's critical quality attribute. Further, changes in higher order structure can impact quality, stability, potency, and efficacy of the product with increased potential for immunogenicity and loss of biological function.
Secondary structure relates to the 3D arrangement of local segments of proteins. The major types of secondary structures in a protein molecule are alpha helices, beta sheets, turns, and random coil. The arrangement of the secondary structure of a protein is conserved and is important for proper folding.
When designing a therapeutic drug, secondary structure plays a significant role in determining proper functioning of the protein of interest. An example of this is the formation of loop structures, a common feature in proteins, that play a significant role in downstream signaling by post-translational modifications or as recognition sites for other proteins. Complementarity determining region (CDR) loops of the heavy and light chain in antibodies are a prominent example of this structure (7,8).
Tertiary structure refers to the 3D arrangement of a single polypeptide chain containing all the functional domains of the monomer. Different secondary elements present in the protein fold into stable globular structures through salt bridges, hydrogen bonds, or disulfide bridge formation (9). For a lot of proteins, the tertiary order is the highest and they are fully functional after achieving this conformation (for example, insulin, granulocyte-colony stimulating factor). However, certain biotherapeutics, such as mAbs, attain their functionality via a specific arrangement of the different monomers (light chain, heavy chain) into a quaternary structure (2,10).
Quality Attributes of Biotherapeutic Products
An inherent amount of heterogeneity is always present in biotherapeutic products in response to variations in the biological processing of the host organism used for production. Controlling manufacturing processes precisely at an intracellular level such that the product at the end of each cycle is identical is a significant task. It has been shown in several recent publications that even innovator products show a wide range of variability in their critical quality attributes over long periods of time, possibly as a result of process improvements, technology transfers, scale-ups, and other such product life-cycle associated activities (11). This variability is of particular importance to the biosimilar industry because the approval of a biosimilar is based on the premise that the biosimilar product is analytically similar to the innovator product and hence a reduced clinical evaluation is warranted (4).
In this context, a major issue is to define how much variability is acceptable (12). This step requires risk assessment and an understanding of the types of product heterogeneity, its origin in the manufacturing process, as well as its effect on product potency and efficacy. Product associated features that have an impact on quality and under which the heterogeneity is assessed and characterized are known as quality attributes (QA). Amongst the numerous QAs, some are critical to effective functioning of the drug as well as its clinical safety and are known as critical quality attributes (CQA). Other attributes or variation that may not directly impact the drug efficacy or its clinical safety, but are important from a process and product consistency point of view are known as key quality attributes (KQA) (13–15). Quality attributes and their potential impact on overall product performance are listed in Table I and discussed in further detail in the subsequent section.


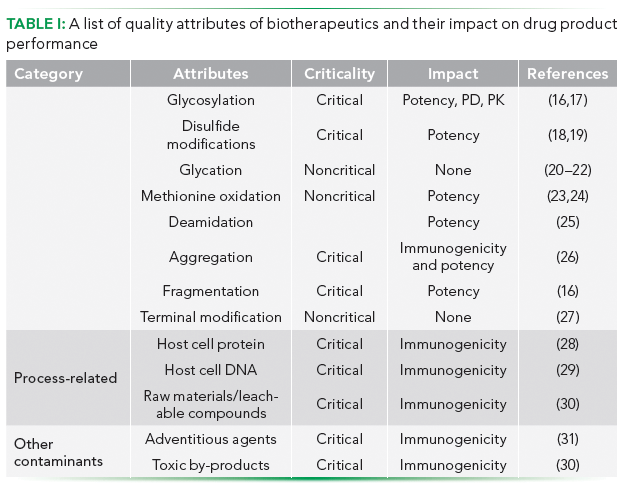
Table I: A list of quality attributes of biotherapeutics and their impact on drug product performance
Post-Translational Modifications
Translation is the process by which ribosomes in a cell's cytoplasm process specific protein-coding gene sequences into proteins. Any modifications that may occur post the release of the protein from the ribosomal assembly come under the category of post-translational modifications (PTMs). Some PTMs are essential for the protein to attain its function and are hence desirable or even essential in the biotherapeutic manufacturing process. PTMs that are essential for function mostly occur inside the cell (in vivo PTMs). Other PTMs might occur outside the cell post lyses (in vitro PTMs), in response to stress, the sample handling, or deliberate addition of the chemical group to improve a certain product attribute (such as PEGylation). Such PTMs are generally undesirable because they add to the pool of protein variants in the formulation (1,32).
Glycosylation
Of the different PTMs that are known to occur in the cell, the most studied PTM relevant to biotherapeutics, especially to mAbs, is glycosylation. This PTM involves the addition of sugar residues to amino acids bearing amino or hydroxyl groups. Glycans (monosaccharides linked glycosidically) are highly versatile in their size (ranging from a few hundred up to several thousand daltons) and in their arrangement (single, linear, or branched).
The glycan profile of a biotherapeutic, especially mAbs can be highly variable owing to the number of possible arrangements of the sugar moieties. Depending on the site of attachment to the polypeptide backbone, glycans can be N-linked or O-linked. Depending on the type and site of glycosylation, the presence or absence of glycosylation can lead to increased stability, reduced aggregation, and changes in potency. A popular example of the negative impact of the absence of glycosylation on activity is interferon-beta (INF-b), where glycosylation imparts a significant increase in protein activity (33–35). In complex molecules such as mAbs, glycosylation of the Fc region has been shown to be essential for FcyII and FcyIII receptor binding (36). Inversely, an absence of fucosylation leads to an increase in FcyIII receptor binding in mAbs (33,37).
Disulfide Bond Formation
Another functionally important PTM with the potential effect on protein stability and efficacy is the disulfide bond formation. Disulfide bonds can form between two cysteine residues within a single polypeptide chain, such as in insulin, or between two proteins to stabilize a complex like immunoglobulins (IgGs). Cysteine residues that are structurally or functionally important for a class of proteins are usually present within the evolutionarily conserved domains of the protein family, such as in human immunoglobulins and cysteine synthase complex in plants. For small molecules, the probability of disulfide bond scrambling is low because of the presence of limited cysteine residues. However, with complex molecules such as the monoclonal antibody complex, the 16 disulfide bonds present are crucial to the stability and hence the downstream efficacy of the complex (18,19).
Glycation
Glycation is the nonenzymatic addition of a monosaccharide to a protein or lipid molecule that usually occurs upon prolonged incubation with reduced sugars. As it is a chemically driven modification, the attachment of monomeric sugar adjacent to a lysine molecule can also occur post administration of the biotherapeutic, during its circulation time in the blood. Therefore, it is critical to ascertain its effects on induced immunogenicity. After they are glycated, proteins can undergo further oxidation to generate a complex set of end products, collectively termed advanced glycation end (AGE) products. These products are involved in a range of pathological conditions like diabetes and osteoarthritis (38,39). In biotherapeutics, studies done so far have shown that induced glycation increases the clearance rate and reduces the circulation time of recombinant IgGs in mice (21). However, this might be specific to the point of glycation on the mAb as well as the mechanism of clearance, as indicated by a study done on the effect of glycation on the Fc region of a mAb where glycation of 0.5 Glc/Lys did not show any significant difference to FcyIII receptor binding (20,21).
Oxidation
Oxidation of a protein is caused because of covalent modification of a protein upon exposure to reactive oxygen species (ROS) or reaction with by-products of oxidative stress. Modifications because of oxidation lead to a range of changes in the protein structure and behaviors such as increased side-chain hydrophilicity, side-chain and backbone fragmentation, aggregation via covalent cross-linking or hydrophobic interactions, protein unfolding and altered conformation, altered interactions with biological partners, and modified turnover (40). In the context of biotherapeutics, oxidation of methionine and tryptophan residues are known to be common degradation pathways, especially for complex molecules such as mAbs. Oxidation of conserved methionine at CH2-CH3 sites (Met256 and Met432) has been shown to cause a loss of FcRn receptor binding resulting in a loss of efficacy (41). Moreover, as the protein A and protein G sites are located close to the CH2-CH3 region, oxidation of Met256 and Met432 has also been shown to cause a decrease in protein A and G binding, thus leading to a loss in manufacturing productivity (23). Because of the above reasons, it is considered a CQA for biotherapeutics and as such is closely monitored with a well-defined, acceptable range for quality tolerance.
Deamidation
Similar to oxidation and glycation, deamidation is a nonenzymatically driven chemical reaction that leads to conversion or removal of an amide functional group in the side chain asparagine (asn) or glutamine (Gln) residues in a protein. Asn is converted to aspartic acid (Asx) or isoaspartic acid and Glu to α- and γ-Glu (42). Deamidation of proteins has been shown to be a contributing factor to aging and several diseases such as celiac disease, urinary tract infection, cataract formation, cancer, and neurodegenerative diseases. The rate of deamidation is affected by physiological conditions around the protein such as pH. The effect of deamidation on protein function is not clear because this modification does not always alter the functionality of a protein. However, cases where an effect on protein function have been reported include glycoprotein CD4 (43), where deamidation at Asn52-Asp53 of CD4 reduced the in vitro biological activity of the product. In the context of biotherapeutics, because of its potential effect on protein turnover and function, it is considered a CQA for biotherapeutic manufacturing (42,44,45).
Aggregation and Fragmentation
All self-associations of proteins into assemblies other than the native HOS come under the umbrella term "aggregates." Aggregates can have a range of species depending upon size (dimer, trimer), type of interaction (covalent, noncovalent), solubility (soluble, nonsoluble), type of formation (ordered and disordered), and mode of formation (reversible, nonreversible) (46). Initiation of aggregation can take place in response to various abiotic (temperature, pH, shear, and pressure) and biotic stresses (immunological state of the host species and genetic predisposition). Because of the multitude of causal agents, predicting aggregation is a challenging task (33).
In therapeutic proteins, the aggregation phenomenon is of particular interest because of its potential for varied immunological responses in humans. Aggregated drug species can elicit formation of antidrug antibodies that bind the drug molecule, thus reducing its efficacy and also activation downstream of immune responses in patients (47). Immune- response pathways, specific to aggregate species, still need to be segregated and understood in detail to predict the type of immune response elicited by a certain aggregate. Instances that have served as learning experiences for the biotherapeutic manufacturing industry include early formulations of intravenous immune globulin (IVIG) and human growth hormone where antibody mediated adverse events were linked with the presence of aggregate species in the formulation (48).
The activity of a protein is a consequence of its sequence. In a multimeric protein such as a mAb, different interacting partners have different functions. Loss of any of the subproteins of the assembly typically leads to loss of the associated function. Fragmentation of protein can occur both enzymatically or nonenzymatically through chemical disruption of the peptide bond. The impact of fragmentation would depend on the specific activity of the cleaved portion. For example, generation of Fab fragments by cleavage at hinge region leads to an altered pharmacokinetic profile. Similarly, dissociation of the variable region responsible for antigen recognition leads to disruption in downstream signaling and activation of associated immune pathways, thus reducing drug efficacy (33).
C- and N-Terminal Modifications
Modifications such as C-terminal lysine residue and N-terminal Gln cyclization are characteristic of the immunoglobulin class of biotherapeutics. The C-terminal of the heavy chain of monoclonal antibody assembly terminates with a lysine that is post-translationally cleaved by endogenous carboxypeptidases. The use of mammalian cells such as Chinese hamster ovary cells is important in this respect because a majority of the product is C-terminal lysine truncated. However, certain variation does exist, which is represented in the basic charge variant profile of the final product. The residual C-terminal lysine is cleaved off post-administration by the patient's endogenous carboxypeptidases. Although this modification is monitored and characterized in the final product, it is not considered as critical for the efficacy and safety of the product (33,49).
Cyclization of the N-terminal (Gln) or Glu to form pyroglutamate (pyroE) and incomplete removal of leader sequence are the two major types of N-terminal modifications of monoclonal antibodies. PyroE formation is a spontaneous event that occurs even during post-drug administration. Studies have shown that the presence of PyroE does not lead to any changes in the antibody structure or affect its receptor binding kinetics. Another form of N-terminal modification is the presence of portions of uncleaved leader peptide sequence. Because of the failure of efficiency in cell cycle machinery, a percentage of the recombinant protein might show the presence of uncleaved leader sequence. Although it contributes to the charge variant species in the final product, it has not been known to cause an immunogenic response in humans. Moreover, studies have shown that the presence of the extra amino acids does not cause any structural change or negatively impact the binding kinetics (50,51).
Process-Related Factors
Process-related impurities consist of contaminants generated by the host cell itself (DNA and host cell proteins), as well as the components of raw materials used throughout the manufacturing process.
Host Cell Impurities
Host cell impurities are additions due to the production of the biotherapeutic in a biologically active cell. Major contaminants in this category are host cell DNA and host cell proteins. During cell lysis, host cell DNA released can carry over into the final product formulation. The risk associated with this contaminant comes from the integration of host cell DNA into the human genome. An early study in which 108 genome equivalents of DNA from a human tumor cell line was injected into nonhuman primates showed no evidence of neoplastic disease, post 8 years of observation. Currently, the World Health Organization (WHO) outlines 10 ng of host cell DNA per dose as the permissible limit (29,33,52).
Other cell components that can carry over into the final formulation are the host cell proteins (HCPs). Given the substantial proteome of hosts used in the biopharma industry, especially the mammalian cell lines such as Chinese hamster ovary cells, this is a potential cause of concern because the presence of certain HCPs in the formulation can cause immunogenic response upon administration. The susceptibility of immunogenic response is seen to be higher for bacterial proteins, probably because of the larger phylogenetic distance between the bacterial systems and primates as opposed to mammalian systems. As per the International Conference on Harmonization (ICH) Q6b, an acceptable range of HCPs in the final formulation is under 100 ppm (53). Immunogenic responses to specific host cell proteins are yet to be studied, but the improvement of purification processes has led to a decrease in immunogenic responses indicating that the presence of HCPs does have some effect in aggravating the immune system (54,55).
Miscellaneous Components
Other adventitious contaminants include viruses, nonhost origin bacterial population, fungal contamination, mycoplasma, endotoxins, and impurities from contaminated raw materials. Sufficient viral clearance needs to be demonstrated for biotherapeutics to be considered safe for commercial use (56).
Summary
With the advent of genetic engineering, it has become possible to engineer and manufacture complex molecules. This capability has revolutionized modern medicine leading to a rapidly evolving segment of biotherapeutics. But as the old adage goes, "with great power comes great responsibility." There is the responsibility to understand the quality attributes of a therapeutic protein product and its clinical efficacy. A better understanding of the product allows for efficient and effective risk assessment. In this regard, implementing scientific practices to define CQAs on a case by case basis is becoming a mandatory, regulatory exercise.
Although a large amount of analytical information is now generated during characterization of a biotherapeutic, gaps exist. There are also technical challenges in generating critical information required to assess the effect of certain modifications on a product's safety and efficacy. For example, isolating individual aggregate species to study their immunogenic effect is a technically challenging task because there is no definitive way of arresting aggregation at a specific stage. Another attribute that has not been completely explored yet is glycosylation. Assessing the individual immunogenic effect and the effect on enzyme kinetics for each of a large number of possible combinations of this modification is currently an interesting hurdle that is yet to be overcome. Many tools are now routinely used for product characterization. As the field grows, innovative technologies need to be used to advance the current knowledge barriers in the characterization of quality attributes for this segment of pharmaceuticals.
References
(1) P.J. Declerck, Generics Biosimilars Initiat. J. 1(1), 13–16 (2012).
(2) S.A. Berkowitz, Analytical Characterization in Biosimilar Drug Prod. Dev. 216, 15–82 (2017).
(3) A. AL-Sabbagh, E. Olech, J.E. McClellan, and C.F. Kirchhoff, Semin. Arthritis Rheum. 45(5), S11–S18 (2016).
(4) A.S. Rathore, Trends Biotechnol. 27(12), 698–705 (2009).
(5) B. Alberts, J. Alexander, J. Lewis, M. Raff, K. Roberts, and P. Walter, in Mol. Biol. Cell. 4th Ed. (Garland Science, New York, 2002).
(6) L.M. Weiner, R. Surana, and S. Wang, Nat. Rev. Immunol. 10(5), 317–327 (2010).
(7) L. Jones and A. McKnight, Biotherapeutics: Recent Developments Using Chemical and Molecular Biology (RSC, Cambridge, 2013), pp. 263–264.
(8) B. North, A. Lehmann, and R.L. Dunbrack, J. Mol. Biol. 406(2), 228–256 (2011).
(9) B.E. Tropp, Molecular Biolgy: Genes to Proteins, 4th Edition (Jones & Bartlett Learning, Burlington, 2012), pp. 27–74.
(10) A. Beck, E. Wagner-Rousset, D. Ayoub, A. Van Dorsselaer, and S. Sanglier-Cianférani, Anal. Chem. 85(2), 715–736 (2013).
(11) L. Halim, V. Brinks, W. Jiskoot, S. Romejin, R. Haselberg, C. Burns, M. Wadhwa, and H. Schellekens, J. Pharm. Sci. 105(2), 542–550 (2016).
(12) S. Fekete, D. Guillarme, P. Sandra, and K. Sandra, Anal. Chem. 88(1), 480–507 (2016).
(13) O. Kwon, J. Joung, Y. Park, C.W. Kim, and S.H. Hong, Biologicals 48, 101–108 (2017).
(14) S.N. Solanke, Indian J. Basic Appl. Med. Res. 3(4), 350–355 (2014).
(15) CMC Biotech Working Group, “A-Mab: A Case Study in Bioprocess Development,” (CASSS, Emeryville, California, 2009).
(16) L. Liu, Protein Cell 9(1), 15–32 (2018).
(17) M.M.C. Van Beers and M. Bardor, Biotechnol. J. 7(12), 1473–1484 (2012).
(18) B. Moritz and J.O. Stracke, Electrophoresis 38(6), 769–785 (2017).
(19) L. Zhang, C.P. Chou, and M. Moo-Young, Biotechnol. Adv. 29(6), 923–929 (2011).
(20) J. Yang, R. Primack, M. Frohn, W. Wang, P. Luan, M.W. Retter, and G.C. Flynn, AAPS J. 17(1), 237–44 (2015).
(21) A.M. Goetze, Y.D. Liu, T. Arroll, L. Chu, and G.C. Flynn, Glycobiology 22(2), 221–234 (2012).
(22) S. Fischer, J. Hoernschemeyer, and H.-C. Mahler, Eur. J. Pharm. Biopharm. 70(1), 42–50 (2008).
(23) G. Gaza-Bulseco, S. Faldu, K. Hurkmans, C. Chumsae, and H. Liu, J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 870(1), 55–62 (2008).
(24) J. Gao, D.H. Yin, Y. Yao, H. Sun, Z. Qin, C. Schöneich, T.D. Williams, and T. Squier, C. Biophys. J. 74(3), 1115–1134 (1998).
(25) M. Haberger, K. Bomans, K. Diepold, M. Hook, J. Gassner, T. Schlothauer, A. Zwick, C. Spick, F.J. Kepert, B. Hienz, M. Wiedmann, H. Beck, P. Metzger, M. MØlhØj, C. Knoblich, U. Grauschopf, D. Reusch, and P. Bulau, MAbs 6(2), 327–39 (2014).
(26) A. Mire-Sluis, B. Cherney, R. Madsen, A. Polozova, A. Rosenberg, H. Smith, T. Arora, and L. Narhi., BioProcess Int. 9(10), 38–43 (2011).
(27) G. Jiang, C. Yu, D.B. Yadav, Z. Hu, A. Amurao, E. Duenas, M. Wong, M. Iverson, K. Zheng, X. Lam, J. Chen, R. Vega, S. Ulufatu, C. Leddy, H, Davis, A. Shen, P.Y. Wong, R. Harris, J.Y. Wang, and D. Li., J. Pharm. Sci. 105(7), 2066–2072 (2016).
(28) V. Jawa, M.K. Joubert, Q. Zhang, M. Deshpande, S. Hapuarachchi, M.P. Hall, and G.C. Flynn, AAPS J. 18(6), 1439–1452 (2016).
(29) D.E. Wierenga, J. Cogan, and J.C. Petricciani, Biologicals 23(3), 221–224 (1995).
(30) W. Wang, A.A. Ignatius, and S.V Thakkar, J. Pharm. Sci. 103(5), 1315–1330 (2014).
(31) J.-P. Gregersen, Vaccine 26(26), 3332–3340 (2008).
(32) G.A. Hernández, Z. Szekanecz, E. Mysler, V.F. Azevedo, R. Guzman, M. Gutierrez, W. Rodriguez, and D. Karteev, Jt. Bone Spine 81(6), 471–477 (2014).
(33) A. Eon-Duval, H. Broly, and R. Gleixner, Biotechnol. Prog. 28(3), 608–622 (2012).
(34) L. Runkel, W. Meier, R.B. Pepinsky, M. Karpusas, A. Whitty, K. Kimball, M. Brickelmaier, C. Muldowney, W. Jones, and S.E. Goelz, Pharm. Res. 15(4), 641–649 (1998).
(35) F. Higel, A. Seidl, F. Sörgel, and W. Friess, Eur. J. Pharm. Biopharm. 100, 94–100 (2016).
(36) J.M. Hayes, E.F.J. Cosgrave, W.B. Struwe, M. Wormald, G.P. Davey, R. Jefferis, and P.M. Rudd, Curr. Top. Microbiol. Immunol. 382, 165–199 (2014).
(37) F. Nimmerjahn and J.V. Ravetch, Nat. Rev. Immunol. 8(1), 34–47 (2008).
(38) H. Liu, “Study of Glycation and Advanced Glycation on a Humanized Monoclonal Antibody” (Doctoral issertation), Retrieved from Open Access Dissertations (2013)
(39) R.A. Saleem, B.R. Affholter, S. Deng, P.C. Campbell, K. Matthies, M.C. Eakin, and A. Wallace, MAbs 7(4), 719–31 (2015).
(40) M.J. Davies, Biochem. J. 473(7), 805–825 (2016).
(41) J. Mo, Q. Yan, C.K. So, T. Soden, M. J. Lewis, and H. Ping, Anal. Chem. 88(19), 9495–9502 (2016).
(42) X. Li, C. Lin, and P.B. O’Connor, Anal. Chem. 82(9), 3606–3615 (2010).
(43) G. Teshima, J. Porter, K. Yim, V. Ling, and A. Guzzetta, Biochemistry 30(16), 3916–3922 (1991).
(44) N.V. Plotnikov, S.K. Singh, J.C. Rouse, and S. Kumar, J. Phys. Chem. B 121(4), 719–730 (2017).
(45) J.R. Lorenzo, L.G. Alonso, and I.E. Sánchez, PLoS One 10(12), (2015).
(46) K.D. Ratanji, J.P. Derrick, R.J. Dearman, and I. Kimber, J. Immunotoxicol. 11(2), 99–109 (2014).
(47) M. Krishna and S.G. Nadler, Front. Immunol. 7, 21 (2016).
(48) A.S. Rosenberg, AAPS J. 8(3), E501–E507 (2006).
(49) F. Torkashvand and B. Vaziri, Iran. Biomed. J. 21(3), 131–141 (2017).
(50) L.A. Khawli, S. Goswami, R. Hutchinson, W.Z. Kwong, J. Yang, X. Wang, Z. Yao, A. Sreedhara, T. Cano, D. Tesar, I. Nijem, D.E. Allison, P.Y. Wong, Y.H. Kao, C. Quan, A. Joshi, R.J. Harris, and P. Motchnik, MAbs 2(6), 613–624 (2010).
(51) H. Liu, G. Ponniah, H.M. Zhang, C. Nowak, A. Neill, N. Gonzalez-Lopez, R. Patel, G. Cheng, A.Z. Kita, and B. Andrien, MAbs 6(5), 1145–1154 (2014).
(52) C.E. Hogwood, D.G. Bracewell, and C.M. Smales, Bioengineered 4(5), 288–291 (2013).
(53) International Conference on Harmonization, Expert Working Group, Specif. Test Proced. Accept. Critreia Biotechnol. Prod. (ICH, Geneva, Switzerland, 1–20, 1999).
(54) A.J. Chirino and A. Mire-Sluis, Nat. Biotechnol. 22(11), 1383–1391 (2004).
(55) B. Sharma, Biotechnol. Adv. 25(3), 325–331 (2007).
(56) D.M. Strauss, T. Cano, H. Delucchi, M. Plancarte, D. Coleman, G.S. Blank, Q. Chen, and B. Yang, Biotechnol. Prog . 26(3), 750–755 (2010).
ABOUT THE AUTHORS


Anurag S. Rathore

Anurag S. Rathore is a professor in the Department of Chemical Engineering at the Indian Institute of Technology in Delhi, India.


Srishti Joshi

Srishti Joshi is a post-doctoral research fellow in the Bioseparations and Bioprocessing Lab, under the tutelage of Professor Anurag. S. Rathore, Department of Chemical Engineering, Indian Institute of Technology, Delhi. She is part of the analytical team, focusing on analytical characterization of biotherapeutics.


Ira S. Krull

Ira S. Krullis a Professor Emeritus with the Department of Chemistry and Chemical Biology at Northeastern University in Boston, Massachusetts, and a member of LCGC's editorial advisory board.
















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


Regulatory Deadlines and Supply Chain Challenges Take Center Stage in Nitrosamine Discussion
April 10th 2025During an LCGC International peer exchange, Aloka Srinivasan, Mayank Bhanti, and Amber Burch discussed the regulatory deadlines and supply chain challenges that come with nitrosamine analysis.

