Analysis of MCPD and Glycidyl Fatty Acid Esters in Refined Plant Oils by Supercritical Fluid Chromatography– High-Resolution Mass Spectrometry
The aim of this study was to develop an efficient method to enable a rapid determination of nine 3-MCPDEs and seven GEs in vegetable oils within a single run employing supercritical fluid chromatography coupled to high-resolution tandem mass spectrometry (SFC–HRMS/MS).
2- and 3-chloropropane-1,2-diol (2- and 3-MCPD) and their fatty acid esters (2- and 3-MCPDEs) together with glycidyl fatty acids esters (GEs) belong to a group of process-induced contaminants. They are formed in high temperature environments and most commonly occur in refined vegetable oils. Since 2021, maximum limits for GEs and 3-MCPDEs have been enforceable in the EU, and the requirement for adequate analytical methods is constantly increasing. The aim of this study was to develop an efficient method to enable a rapid determination of nine 3-MCPDEs and seven GEs in vegetable oils within a single run employing supercritical fluid chromatography coupled to high-resolution tandem mass spectrometry (SFC–HRMS/MS). Contrary to a routine gas chromatography mass spectrometry method aimed at determination of the total MCPD pool, the sample throughput when using SFC–HRMS/MS was greatly increased, as the “dilute-and shoot” approach did not require any hydrolysis and derivatization. Additionally, the pattern of natural MCPDEs was characterized. The performance characteristics of this new method met the criteria required by Commision Regulation 2019/2093 for all target analytes.
2- and 3-chloropropane-1,2-diol (2- and 3-MCPD) together with glycidol belong to a group of process‑induced chemical contaminants of edible oils. Chloropropanediols and glycidol are derived from glycerol, and, similar to glycerol, they occur mainly in their ester‑bound form as 2- and 3-chloropropanediol mono- and diesters (2- and 3-MCPDEs) and glycidyl esters (GEs). The profile of bound fatty acids usually corresponds to that in the respective plant oil (1–2). Numerous studies have shown that they are not present in crude oils and occur only after high temperature treatment, such as during oil deodorization (2–6).
The International Agency for Research on Cancer classifies 3-MCPD as possibly carcinogenic to humans (Group 2B) and glycidol as probably carcinogenic to humans (Group 2A). Toxicity of 2-MCPD and its esters requires further investigation and is yet to be classified in terms of its carcinogenicity and dose‑response assessment (7–12). Although most food-borne 3-MCPD occurs in its bound form (approximately 85% diesters and 15% monoesters), previous studies have shown that its free form is effectively released in vivo by enzymatic hydrolysis in the gastrointestinal tract (13). For the purpose of risk assessment, ingested bound 3-MCPD is usually considered as molar equivalent to its free form. In 2016, the Contaminants in the Food Chain (CONTAM) panel of the European Food Safety Authority (EFSA) assessed known risks associated with 2- and 3-MCPD and established a tolerable daily intake (TDI) of 2 μg/kg body weight per day for 3-MCPD and its fatty acid esters, which are expressed as MCPD equivalents. EFSA further concluded that adequate toxicological data for free and bound 2-MCPD are currently not available (9,11,13–16).
Following these findings, the Directorate-General for Health and Food Safety (DG SANTE) proposed maximum limits for GEs in several food commodities. These have been further expanded to include 3-MCPDEs, and since 2021, these limits are enforceable in the European Union as a part of Commision Regulation 2020/1322 (17). The requirements for adequate analytical methods are therefore constantly increasing.
There are two main approaches to MCPDEs and GEs analysis: either directly determine their content in intact ester forms or release them all into their free forms and determine them indirectly. The majority of currently used methods are based on the indirect approach, which involves analysis of 2- and 3-MCPD and glycidol after hydrolysis (either chemical or enzymatic) and conversion to volatile derivates, followed by gas chromatography–mass spectrometry (GC–MS) analysis. Their main advantages lie in lower analytical standard requirements and applicability in a wider range of food categories (18–19). However, the accuracy and repeatability of these methods was disputed due to the harsh chemical conditions required during sample preparation, which can interfere with MCPD and glycidol content (20). In combination with often lengthy sample preparation of indirect methods, direct approaches are often promoted as a more suitable alternative (18).
Direct methods have the obvious advantage in providing profiles of individual MCPDEs and GEs. The fatty acid composition not only offers a better insight in MCPDEs and GEs formation and subsequent development of mitigation strategies but it is also suggested that individual esters might differ in their overall toxicity. On the other hand, a higher number of analytical standards and often more expensive instrumentation are required (18,21).
In most cases the direct method workflow involves liquid chromatography–mass spectrometry (LC–MS) analysis, either in a tandem mass spectrometry (MS/MS) or high-resolution mass spectrometry (HRMS) arrangement. As the vast majority of these systems operate in reversed‑phase mode, the major issue then arises in the determination of MCPDEs and GEs in oily matrices. The presence of large amounts of acylglycerols, particularly triacylglyceroles (TAG), negatively affect the analysis. Therefore, most published methods include a sample purification step, usually by the use of solid-phase extraction (SPE), often in multiple stages (reversed‑phase SPE, followed by normal‑phase SPE), prior to LC–MS (4,18–19,22–23). These additional steps have a significant impact on sample throughput and the overall cost of these methods.
As a possible solution, methods based on supercritical fluid chromatography (SFC) for direct determination of MCPDEs have been published in recent years (24–25). The mobile phase of choice for modern SFC systems is supercritical CO2, which displays characteristics similar to pentane or hexane, making it suitable for the separation of lipophilic compounds (26). Indeed, SFC coupled with MS/MS or HRMS was shown to provide rapid, high-throughput “dilute and shoot” methods for MCPDEs analysis. Obtained method performance characteristics indicate a potential of this approach to be used for quantification of these processing contaminants. Furthermore, the use of CO2 instead of commonly used organic solvent as the mobile phase is considered green (24–25).
The aim of this study was to develop and optimize a method enabling a rapid, simultaneous determination of not only various intact esters of 3-MCPDEs but also GEs, in various vegetable oils and fats. The use of supercritical fluid chromatography coupled with high resolution mass spectrometry (SFC–HRMS) was addressed as a analytical challenge. Achieving criteria for performance characteristics set in European legislation (Commision Regulation 2019/2093 [27]) for methods targeting 3-MCPDEs and GE in plants was the ultimate objective.
Materials and Methods
Chemicals: Standards of racemic 3-MCPD fatty acids diesters were as follows: 1,2-dipalmitoyl-3-chloropropane-1,2-diol (PP-3-MCPD); 1-palmitoyl-2-linoleoyl-3-chloropropanediol (PL-3-MCPD); 1-palmitoyl-2-oleoyl-3-chloropropanediol (PO-3-MCPD); 1-palmitoyl-2-stearoyl-3-chloropropanediol (PSt-3-MCPD); 1,2-dilinoleoyl-3-chloropropanediol (LL-3-MCPD); 1,2-dioleoyl-3-chloropropanediol (OO-3-MCPD); 1-oleoyl-2-linoleoyl-3-chloropropanediol (OL-3-MCPD); 1-oleoyl-2-stearoyl-3-chloropropanediol (OSt-3-MCPD); 1,2-distearoyl-3-chloropropanediol (StSt-3-MCPD). Available glycidyl esters (GEs) with fatty acids were: glycidyl stearate (G-St); glycidyl palmitate (G-P); glycidyl laurate (G-La); glycidyl myristate (G-M); glycidyl linoleate (G-L); glycidyl linolenate (G-Ln); and glycidyl oleate (G-O). All of these analytical standards were purchased from Toronto Research Chemicals (the declared purity was ≥ 98%). HPLC-grade ethyl acetate, n-hexane, 2-propanol, and methanol were purchased from Honeywell. Deionized water was supplied by the Millipore (Milli - Q) purification system from Merck. Ammonium acetate for mass spectrometry was obtained from Merck. Nitrogen and carbon dioxide (purity 4.0) were purchased from SIAD. Two sets of stock standard solutions—one containing all MCPDEs standards, the other containing all GE—were prepared in ethyl acetate at 1 μg/mL, labelled MCPDE_1, GE_1, and 10 μg/mL labelled MCPDE_10 and GE_10.
Samples: Refined vegetable oil samples (rapeseed and sunflower; n = 2 and n = 4, respectively) purchased at a local retail market were used for analyte screening and subsequent method validation.
Sample Preparation:
Samples for Target Analytes Screening: A 0.5-g measure of oil was accurately weighted into a 5-mL volumetric flask and the volume adjusted by n-hexane. Two 1-mL aliquots of obtained oil solution (100 mg of oil/mL) were then transferred into 2-mL glass vials for SFC–MS analysis.
Samples for Method Optimization and Validation: A set of 2-mL vials was pre‑loaded with either 15, 20, or 25 μL of both MCPDE_1 and GE_1, or 5 or 10 μL of both MCPDE_10 and GE_10. Vials were then dried in a block heater at 30 °C under a gentle stream of nitrogen. The residue was diluted in 1 mL of hexane to obtain “solvent standard”, or in 1 mL of previously prepared oil solution to get “matrix-matched standard” containing 100 mg of oil/mL (for oil solution preparation, see section above), capped, thoroughly shaken, and subsequently subjected to SFC–MS analysis.
SFC–HRMS Analysis: MCPDEs and GEs analysis was performed using an Aquity UltraPerformance Convergence Chromatography (UPC2) system coupled with Synapt G2-Si, which was equipped with ZSpray electrospray ion source with a hybrid quadrupole–travelling wave ion mobility–time-of-flight mass spectrometer (Waters). A make-up solvent was delivered by a HPLC 515 pump (Waters) and mixed with column effluent in a two T-piece coupling. The combined flow was then divided to an active atmospheric back pressure regulator (ABPR) module and the mass spectrometer. The best SFC–MS conditions found in our study were as follows.
SFC Conditions: Column: 100 × 3 mm, 1.8-μm Viridis BEH (Waters); mobile phase composed of supercritical CO2 with 99:1 (v/v) methanol–water mixture as a co‑solvent containing 30 mM of ammonium acetate as a modifier at a constant flow rate of 1.8 mL/min; the co-solvent followed a linear gradient curve: 0 min, 0.5% -> 4 min, 5% -> 8 min, 45% -> 10 min, 45% -> 11 min, 0.5% -> 12 min, 0.5%; column temperature: 70 °C; ABPR pressure: 13.79 MPa (2000 psi); injection volume: 4 μL; injector needle was washed with a 7:3 (v/v) n-hexane–2-propanol mixture and pure 2-propanol after each injection.
MS Ion Source Conditions: As a make‑up solvent, a mixture of 99:1 (v/v) methanol–water was used at a constant flow rate of 0.3 mL/min. A positive-mode electrospray ionization (ESI+) in resolution mode was used at the following settings of tuning parameters: capillary voltage: +4.5 kV; sampling cone: 40 V; source offset: 80 V; source temperature: 120 °C; desolvatation temperature: 350 °C; cone gas flow: 50 L/h; desolvatation gas flow: 800 L/h; nebulizer pressure: 4 bar.
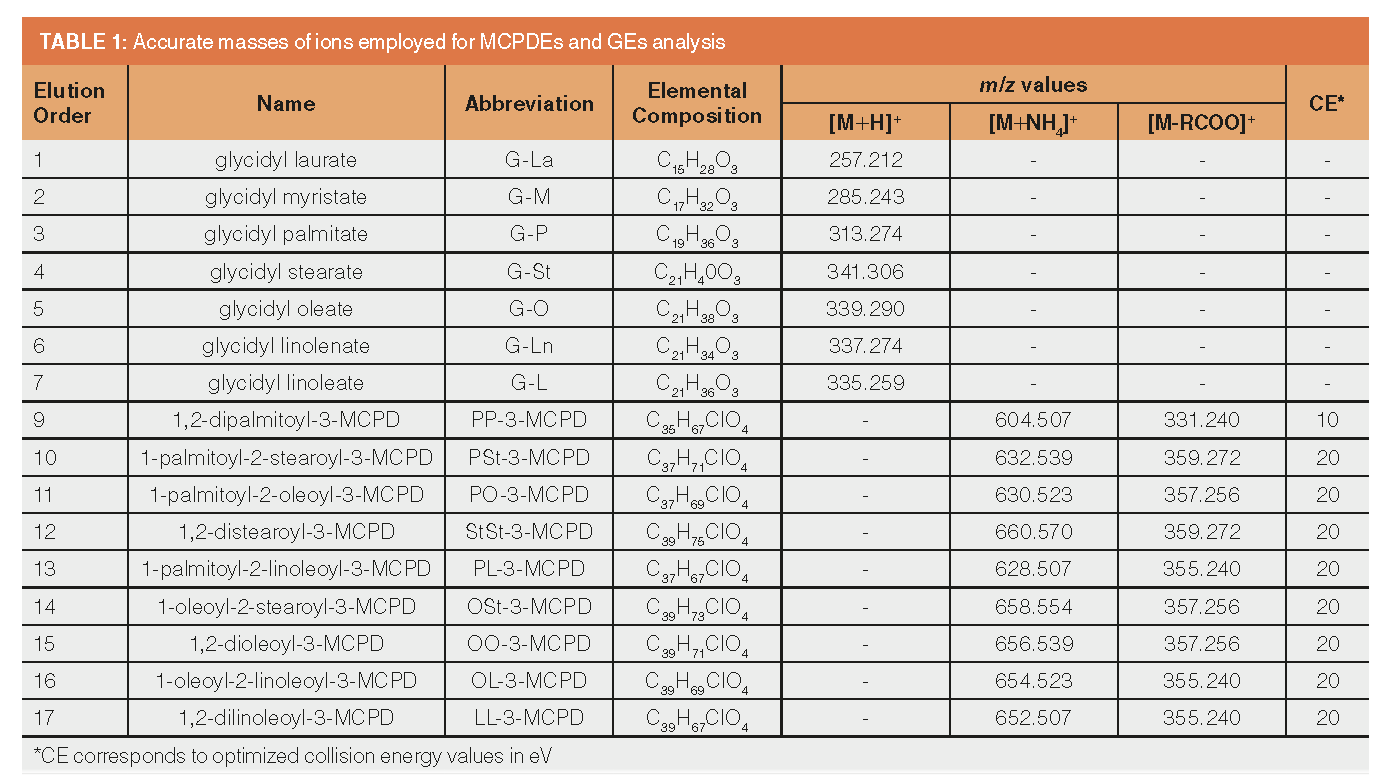
MS Acquisition Conditions: MS data were acquired (i) in HRMS mode for SFC method development and ionization optimization, and (ii) in combined HRMS/MS mode for collision energy optimization and final method validation. The HRMS mode monitored ammonium adducts [M + NH4]+ of MCPDEs and protonated molecules [M + H]+ of GEs. The HRMS/MS method used the [M + NH4]+ adducts of MCPDEs as precursor ions and monitored characteristic decarboxylated ions [M - RCOO]+. The m/z values of characteristic ions are summarized in Table 1. The (i) HRMS data were acquired in m/z range 100–1000. For the (ii) combined HRMS/MS method, the HRMS function mass range was narrowed to m/z 250–400, and for the MS/MS functions for individual MCPDEs to m/z 300–400. MS/MS fragmentation was performed on a transfer cell.
Method Optimization: For method optimization, solutions of MCPDEs and GEs 100 ng/mL (for each analyte) in n-hexane and a spiked oil sample at the same concentration level (corresponds to 1000 μg of each ester/kg of oil sample) were prepared (see Samples for method optimization and validation), both in two replicates, and analyzed with SFC–MS under various conditions. The underlined values represent default method conditions. The outcome of modifications were evaluated based on average (mean of parallel measurements) percentage of response differences of [M + NH4]+ MCPD adducts and [M + H]+ GEs adducts compared to the default settings. Each time, the best performing conditions were used for further steps.
SFC Method Optimization: SFC method parameters subjected to optimization included the following: co-solvent gradient (5% 30 mM of ammonium acetate in 99:1 (v/v) methanol–water co-solvent at 4/5/6/7 min), ABPR pressure (1600/1800/2000/2200 psi), and make-up solvent composition (pure methanol/99:1 [v/v] methanol–water/30 mM of ammonium acetate in 99:1 [v/v] methanol–water).
MS Method Optimization: MS method parameters subjected to optimization included the following: ion source capillary voltage (1.0/1.5/2.0/2.5/3.0/ 3.5/4.0/4.5 kV), ion source temperature (100/120/150 °C), and collision energy for MS/MS analysis (0/10/20/30/40/50 eV).
Method Validation: To validate the previously optimized procedure, a standard addition method (SAM) approach utilizing spiked samples was chosen. Six parallel sets of selected oil solutions were spiked with MCPDEs and GEs stock standard solutions at concentration levels 30, 40, 50, 100, and 200 ng/mL (see Samples for method optimization and validation), which correspond to 300, 400, 500, 1000, and 2000 μg of each ester/kg of oil. Oil solutions without added MCPDEs or GEs (matrix blank samples) were also prepared in six parallels (see Samples for target analytes screening). All samples were then analyzed by SFC–MS. SAM calibration equations were expressed as y = ax + b by the least square method, where a is the slope of the curve and b is the intercept of the y-axis. Analyte concentrations were calculated as a negative horizontal axis intercept (-b/a). Method performance characteristics represented by linearities, limits of quantification (LOQ), repeatabilities, recovery rates, and matrix effects were characterized and assessed against the criteria specified in Commision Regulation 2019/2093 regarding performance criteria for methods of analysis for 3-MCPDEs and GEs. Method linearity was expressed as coefficient of determination (R2) of SAM calibration equations. Recovery rates were evaluated by comparing the average theoretical and determined concentration in parallel sets. Repeatability was expressed as the relative standard deviation (RSD) between determined concentrations in parallel sets. The relative matrix effects were calculated as average ratios between SAM calibration equation slope (a) and solvent calibration equation slope (a´) of equivalent concentration levels: a/a´.
Results and Discussion
SFC Method Optimization Results: As stated above, SFC–MS is a powerful tool for analysis of lipophilic compounds. For fast separation of target analytes from TAGs, the major matrix “interfering” components in refined oil, an ethylene bridged hybrid silica phase column with medium silanol activity was selected. Under optimized conditions, the “dilute and shoot” approach was enabled and both MCPDEs and GEs, which are more hydrophobic than TAGs, eluted at lower retention times and did not suffer from massive matrix effects. This is well outlined in Figure 1, where extracted ion chromatograms (EIC) of MCPD [M + NH4]+ and GEs [M + H]+ adduct ions (for m/z values see Table 1) in spiked rapeseed oil sample are shown, compared to EIC of selected m/z values calculated for [M + NH4]+ TAG adducts. EIC of displayed TAG peaks were selected from species naturally occurring in oil samples to have as low a retention time as possible, to mark the beginning of TAG elution zone. Since the analytes of interest in this study are eluted in the first 2 min, the initial mobile phase composition was lowered from the previously used 1% of co-solvent (30 mM of ammonium acetate in 99:1 [v/v] methanol–water) to 0.5%, and the flow rate decreased from 1.9 to 1.8 mL/min. This allowed for efficient separation of MCPDEs and GEs from the TAG matrix. The main contribution to matrix effect was therefore attributed to more lipophilic compounds, tentatively identified as [M + NH4]+ and [M - H2O + H]+ adducts of phytosterol and retinol esters (see Figure 1). The modified method was used as a basis for further optimization with regards to obtaining maximal responses and minimal LOQs.


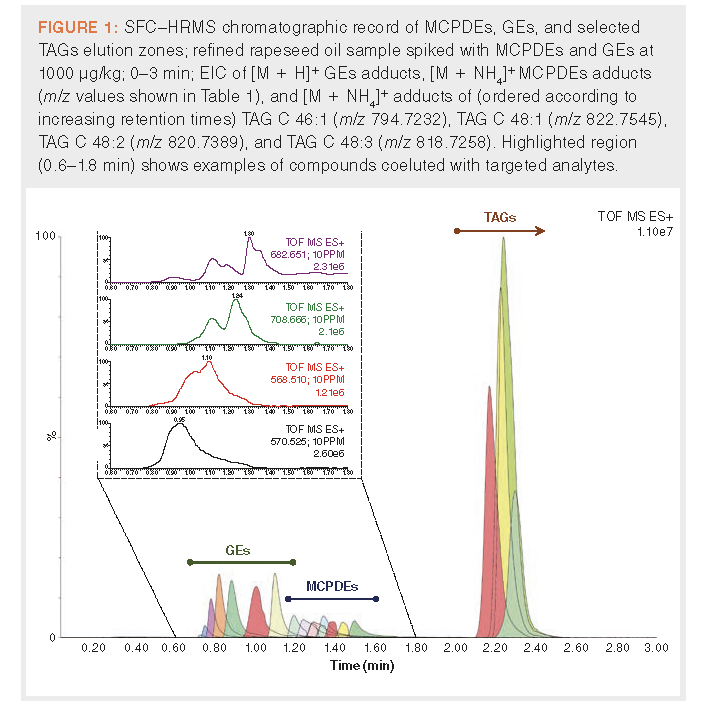
The subject of gradient optimization was the initial slope from 0.5% to 5% of co-solvent at 6 min. The largest increase of responses, measured in solvent standards (100 ng/mL), was observed with the steepest tested gradient (5% co-solvent at 4 min) and was on average 5.1%, with values ranging from +29.7% for G-M to -12.0% for StSt-3-MCPD. However, six out of nine MCPDEs showed a decrease in response. Considering that targeted MCPDEs ions are less intensive when analyzing solution with the same concentration than GEs, and the other two tested gradient settings performed on average worse, the default gradient was kept unchanged.
In the next phase, the ABPR pressure was optimized. Its higher values (and therefore higher CO2 density) generally improve elution strength of supercritical CO2, resulting in narrower and higher peaks. On the other hand, it can also adversely impact the ABPR/MS split ratio during solvent exchange. Nevertheless, in this case, the effect was only marginal, with the best result being an average 2.8% response increase (mean of all analytes) at 2000 psi.
Another factor influencing analyte responses is the make-up solvent composition. From the original make-up solvent (99:1 [v/v] methanol–water), two different approaches were tested. The first attempt was based on excluding water in addition to methanol. It was assumed that, thanks to the increased solubility of relatively nonpolar target analytes in make-up solvent, the ABPR/MS split ratio would be improved. The second alternative involved modifying the existing make-up solvent with the addition of 30 mM ammonium acetate to support [M + NH4]+ formation in the ion source, thus increasing MCPDEs responses. A rapeseed oil solution spiked at 100 ng/mL (1000 μg/kg) was used for this purpose to take into consideration potential matrix effects in this step. Unfortunately, the use of both modified make-up solvents resulted in reduced responses for all analytes, and therefore the original make-up solvent was kept unchanged. A mention should be made that the SFC method presented here resulted in 2- and 3-MCPDEs coelution and, therefore, to some extent 3-MCPDE signals might be increased by 2-MCPDEs.
MS Method Optimization Results: Further method development steps were focused on analyte ionization. The source voltage was optimized on 100 ng/mL solvent standard. Compared to the default method with ionization voltage setting 3.5 kV, its increase to 4.5 kV resulted (with the exception of OL-3-MCPD) in some increase of analyte signal intensities, a maximum of 16.5% in the case of G-P.
MS/MS Method for Analytes Quantification: When targeting individual MCPDEs in chromatographically only partly resolved mixtures (which was the case for the developed high-throughput method), a quantification issue might occur. Like other organochlorine compounds, MCPDEs contain in their isotopic profile a relatively intensive M+2 ion, which may, supposing partial coelution, interfere with targeted ions of other analytes. For instance: the difference in theoretical m/z value 632.5385 of PSt-3-MCPD ion [M + NH4]+ and m/z 632.5220, calculated for M+2 isotope of PO-3-MCPD [M + NH4]+ is only Δm/z = 0.0165. At a given instrument resolution (24,000–30,000 FWHM in “resolution” mode), for the m/z value 632.5385 ions in the range 632.5596–632.5174 cannot be resolved. To overcome this issue and to simultaneously improve on selectivity for MCPDEs, a MS/MS method was designed.
As described in Table 1, suitable product ions were identified. These ions correspond to decarboxylated [M - RCOO]+ fragments of [M + NH4]+ precursors, in accordance with the available literature (24–25). Transfer cell collision energy was selected for each analyte based on responses obtained with each setting on 100 ng/mL hexane MCPDEs solution. With the exception of PP-3-MCPD (at 10 eV), the best results were achieved at 20 eV. An example MS/MS chromatographic record of rapeseed oil spiked with MCPDEs is shown in Figure 2. The resulting optimized method was subsequently validated.


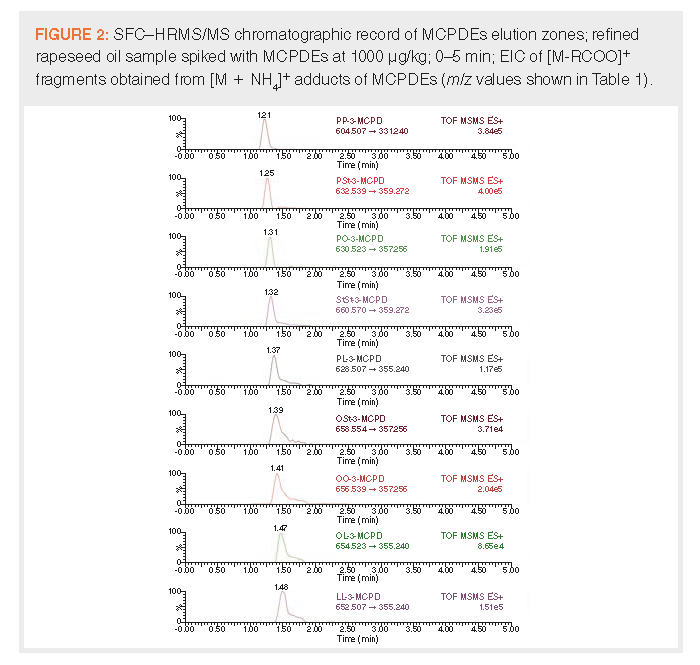
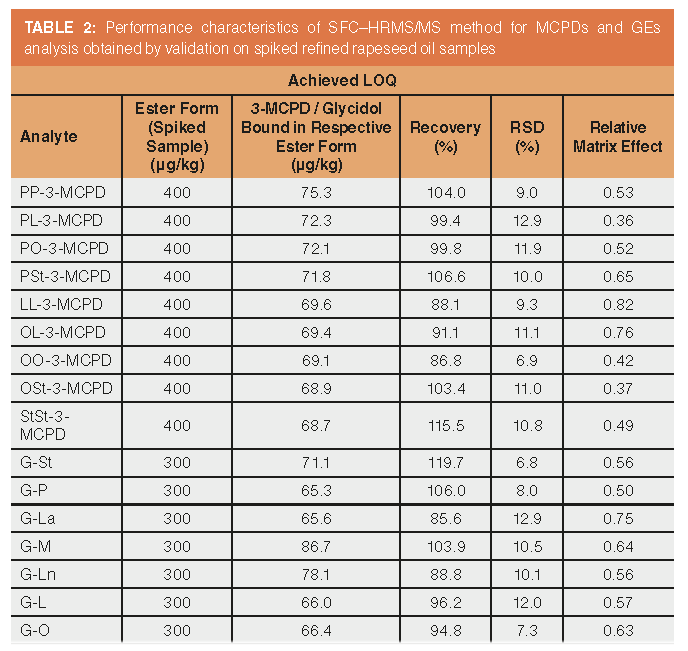
Validation Results: Commision Regulation 2019/2093 specifies method performance criteria that have to be achieved when controlling the regulated levels of 3-MCPDEs and GEs in vegetable oils. These include a recovery rate of 70–125%, LOQ ≤ 100 μg of ester bound 3-MCPD or glycidol/kg of oil, and repeatability expressed as RSD derived from the modified Horwitz equation. LOQ of 100 μg of ester bound 3-MCPD or glycidol/kg of oil corresponds to 582–531 μg of MCPDEs/kg and 346–460 μg of GE/kg, depending on the particular ester form. At such concentration levels, the required repeatability equates to RSD ≤ 14.52%.
Samples of refined vegetable (rapeseed and sunflower, see Samples for target analytes screening) oils were screened for the presence of MCPDEs and GE. The rapeseed oil sample (labelled as “soft refined”) with the lowest contaminants content (G-O, G-L, and G-Ln were detected at estimated concentrations 5, 17, and 24 μg of bound glycidol/kg of oil, respectively) was selected as the blank matrix for SAM method validation (see Method validation). The responses of analytes occurring in such “blank” samples were subtracted from all concentrations determined in spiked samples. Achieved validation parameters are summarized in Table 2. LOQ for each analyte is expressed in μg of ester forms/kg of oil and equivalent μg of ester bound 3-MCPD or glycidol/kg of oil, and represent the lowest concentration of spiked oil sample at which acceptable recovery and RSD were achieved. Obtained LOQs were low enough, ranging between 65–87% of the proposed maximum limit—100 μg of ester bound 3-MCPD or glycidol/kg. Method linearity of SAM calibration equations R2 < 0.99 was achieved for all studied compounds at all spiking levels. All the validation parameters met the required performance criteria.

Conclusion
This study presents a newly developed and validated high-throughput “dilute and shoot” analytical approach for determination of nine esters of 3-MCPD and seven esters of glycerol in refined vegetable oil using SFC–HRMS/MS. The key advantages of the described method, in comparison with conventionally used procedures, consist in minimal sample preparation requirements and no need for chemical modification of analytes or sample clean‑up prior to instrumental analysis. The green nature associated with a minimized solvent consumption is another benefit.
Selected SFC and HRMS conditions, namely co-solvent gradient, ABPR pressure, make-up solvent composition, ion source voltage, and temperature, were optimized to achieve the lowest LOQs and other method performance characteristics. As all of them meet the criteria of European legislation (Commision Regulation 2019/2093), the method is suitable for an official control of both groups of processing contaminants of plant oils.
Acknowledgement
This work was financially supported by METROFOOD-CZ research infrastructure project (MEYS Grant No: LM2018100), including access to its facilities, by the “Operational Programme Prague – Competitiveness” (CZ.2.16/3.1.00/21537 and CZ.2.16/3.1.00/24503) and the “National Programme of Sustainability I” - NPU I LO1601 and by the Czech Republic National Agency for Agricultural Research (Project no.QJ1530272). The support from the grant of specific university research—grant No. A1_FPBT_2022_005 and A2_FPBT_2022_073—are also gratefully acknowledged.
References
- C. Crews, A. Chiodini, M. Granvogl, C. Hamlet, K. Hrnčiřík, J. Kuhlmann, A. Lampen, G. Scholz, R. Weisshaar, T. Wenzl, P.R. Jasti, and W. Seefelder, Food Additives & Contaminants: Part A 30(1), 11–45 (2013).
- T.G. Albuquerque, H.S. Costa, M.A. Silva, and M.B.P.P. Oliveira, Trends in Food Science & Technology 105, 494–514 (2019).
- R. Tivanello, M. Capristo, E. Vicente, R. Ferrari, K. Sampaio, and A. Arisseto, J. Food. Sci. 85(7), 2255–2260 (2020).
- J.K. Beekman, K. Grassi, and S. MacMahon, Food Addit. Contam. Part A Chem. Anal. Control Exposure Risk Assess. 37(3), 374–390 (2020).
- S. MacMahon, E. Mazzola, T.H. Begley, and G.W. Diachenko, J. Agric. Food Chem. 61(20), 4737–4747 (2013).
- B.D. Craft, K. Nagy, L. Sandoz, and F. Destaillats, Food Addit. Contam. Part A Chem. Anal. Control Exposure Risk Assess. 29(3), 354–361 (2012).
- E. Bognár, G. Hellner, A. Radnóti, L. Somogyi, and Z. Kemény, Period. Polytech. Chem. Eng. 64(4), 523–529 (2020).
- S. Andres, K.E. Appel, and A. Lampen, Food Chem. Toxicol. 58, 467–78 (2013).
- EFSA, EFSA Journal 14(5), e04426 (2016).
- IARC, 77, 469–486 (2000).
- IARC, 101, 9–549 (2013).
- Z. Zhang, P. Yang, B. Gao, G. Huang, M. Liu, and L.L. Yu, J. Agric. Food Chem. 67(13), 3789–3795 (2019).
- K. Abraham, K.E. Appel, E. Berger-Preiss, E. Apel, S. Gerling, H. Mielke, O. Creutzenberg, and A. Lampen, Arch. Toxicol. 87(4), 649–659 (2013).
- E. Barocelli, A. Corradi, A. Mutti, and P.G. Petronini, EFSA Supporting Publications 8(9), 187E (2011).
- EFSA, EFSA Journal 16(1), e05083 (2018).
- M.E. Mossoba, M.S.T. Mapa, M. Araujo, Y. Zhao, B. Flannery, T. Flynn, J. Sprando, P. Wiesenfeld, and R.L. Sprando, Cell Biology and Toxicology 36(3), 209–221 (2020).
- European Commision, Commission Regulation (EU) 2020/1322 of 23 September 2020 amending Regulation (EC) No 1881/2006 as regards maximum levels of 3-monochloropropanediol (3-MCPD), 3-MCPD fatty acid esters and glycidyl fatty acid esters in certain foods, 32020R1322, 5 (2020).
- B.Y. Gao, Y.F. Li, G.R. Huang, and L.L. Yu, Fatty Acid Esters of 3-Monochloropropanediol: A Review (Annual Reviews, Palo Alto, USA, Vol. 10, 2019), pp. 259–284.
- M. Dubois, A. Tarres, T. Goldmann, A.M. Empl, A. Donaubauer, and W. Seefelder, Journal of Chromatography A 1236, 189–201 (2012).
- T.D. Haines, K.J. Adlaf, R.M. Pierceall, I. Lee, P. Venkitasubramanian, and M.W. Collison, JAOCS J. Am. Oil Chem. Soc. 88(1), 1–14 (2011).
- M. Liu, J. Liu, Y. Wu, B. Gao, P. Wu, H. Shi, X. Sun, H. Huang, T.T. Wang, and L. Yu, Journal of the Science of Food and Agriculture 97(3), 841–848 (2017).
- J.L. Hidalgo-Ruiz, R. Romero-Gonzalez, J.L.M. Vidal, and A.G. Frenich, Journal of Chromatography A 1639, 9 (2021).
- W.-w. Cheng, G.-q. Liu, L.-q. Wang, and Z.-s. Liu, Comprehensive Reviews in Food Science and Food Safety 16(2), 263–281 (2017).
- K. Hori, A. Matsubara, T. Uchikata, K. Tsumura, E. Fukusaki, and T. Bamba, Journal of Chromatography A 1250, 99–104 (2012).
- F. Jumaah, R. Jędrkiewicz, J. Gromadzka, J. Namieśnik, S. Essén, C. Turner, and M. Sandahl, J. Agric. Food Chem. 65(37), 8220–8228 (2017).
- L. Miller, J. Pinkston, and L. Taylor, Modern Supercritical Fluid Chromatography: Carbon Dioxide Containing Mobile Phases (John Wiley & Sons, Inc., 2019).
- European Commision, Commission Implementing Regulation (EU) 2019/2093 of 29 November 2019 amending Regulation (EC) No 333/2007 as regards the analysis of 3-monochloropropane-1,2-diol (3-MCPD) fatty acid esters, glycidyl fatty acid esters, perchlorate and acrylamide, 32019R2093, 6 (2019).
Tomas Kourimsky is a PhD student in the Department of Food Analysis and Nutrition, UCT Prague. His research is focused on the analysis of MCPD and its esters employing GC–MS/MS and SFC–HRMS.
Vojtech Hrbek is a research assistant in the Department of Food Analysis and Nutrition, UCT Prague. His main research interests include (but are not limited to) application of (high-resolution) mass spectrometry in the analysis of bioactive compounds and contaminants in food.
Martin Steidl is an M.Sc. student in the Department of Food Analysis and Nutrition, UCT Prague. He is a member of a research team engaged in development and optimization of SFC–HRMS methods.
Jana Hajšlová is a professor at UCT Prague. She is the head of ISO 17025/2018 accredited laboratory and also heads a research group concerned with separation science in the field of food analysis.










New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.


Analytical Challenges in Measuring Migration from Food Contact Materials
November 2nd 2015Food contact materials contain low molecular weight additives and processing aids which can migrate into foods leading to trace levels of contamination. Food safety is ensured through regulations, comprising compositional controls and migration limits, which present a significant analytical challenge to the food industry to ensure compliance and demonstrate due diligence. Of the various analytical approaches, LC-MS/MS has proved to be an essential tool in monitoring migration of target compounds into foods, and more sophisticated approaches such as LC-high resolution MS (Orbitrap) are being increasingly used for untargeted analysis to monitor non-intentionally added substances. This podcast will provide an overview to this area, illustrated with various applications showing current approaches being employed.
 Headquarters complex in Washington, DC. | Image Credit: © Tada Images - stock.adobe.com")




