Quantitative Analysis of PFAS in Milk, Infant Formula, and Related Ingredients Using LC–MS
Per- and polyfluoroalkyl substances (PFAS) are a large group of anthropogenic chemicals that have been applied in a wide range of industrial, commercial, and domestic products since the 1950s. Because of their toxicity, persistence, and bioaccumulation potential, PFAS have become global environmental pollutants. Besides the environment, the food chain represents another source of exposure, and the risk to consumers related to the presence of PFAS in foods has recently become of increased interest. In this respect, whole milk, infant formula, and ingredients used in infant formula production represent important foodstuffs that require sensitive methods with reporting limits at low parts per billion levels or lower for multiple PFAS. This article summarizes optimization experiments and validation of a complete workflow, including sample preparation and a liquid chromatography–tandem mass spectrometry (LC–MS/MS) method for determination of sixteen priority PFAS analytes listed by the U.S. Food and Drug Administration (FDA).
There are several ways humans can be exposed to per- and polyfluoroalkyl substances (PFAS). These include exposure during normal use of a wide range of commercial consumer products that have been treated with fluoropolymers or PFAS—for example, to make them non-stick or water- or stain-repellent. Drinking water can also be a source of exposure in locations where these chemicals have contaminated water supplies. Finally, humans can be exposed to these chemicals through contaminated food. When it comes to contamination of food with PFAS, there are two main processes to consider. Bioaccumulation of PFAS in both aquatic and terrestrial food chains is a result of use of contaminated water and soil for growing food. Additionally, contamination of finished products and ingredients may occur due to the use of PFAS containing materials for food packaging and food processing. Presence of PFAS in food and related health risk to consumers has become of increased concern to many regulatory agencies worldwide that have initiated monitoring programs. Besides various foods of animal and plant origin, infant formula and baby food represent another important food commodity of concern regarding the vulnerability of infants and toddlers (1,2).
Rapid, sensitive, and reliable analytical techniques are required for the monitoring of PFAS in the above matrices. Liquid chromatography coupled to tandem quadrupole mass spectrometry (LC–MS/MS) with electrospray ionization (ESI) operated in negative ionization mode has been the instrumental technique most frequently used for the analysis of PFAS in foods, as well as in other biotic matrices (3).
In this study, an LC–MS/MS assay based on the U.S. FDA method C-010.01 was optimized and validated for the analysis of sixteen PFAS compounds in whole milk, ready-to-feed (RTF) and powdered infant formula, milk powder, soybean oil, and maltodextrin matrices (4).
Experimental
Standards, Reagents and Samples
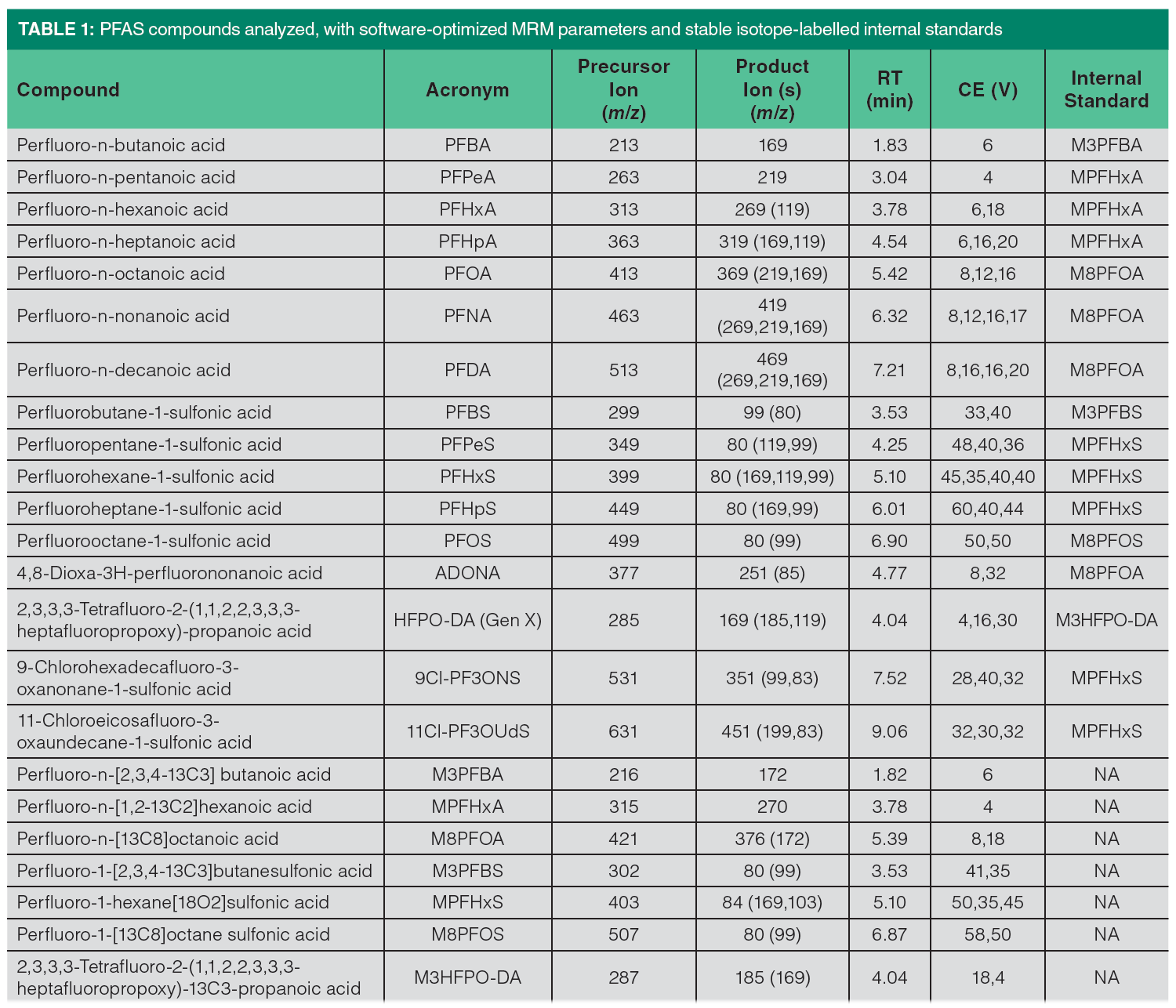
The target PFAS and the stable isotope-labeled internal standards employed in this study are listed in Table 1. The PFAS standards, including isotopically labeled analogs, were purchased from Wellington Laboratories. Formic acid (99%, for analysis) was from Thermo Scientific. LC–MS grade ammonium formate, acetonitrile, and methanol and were from MilliporeSigma. LC–MS grade isopropanol and HPLC grade hexane were from Thermo Fisher Scientific. Ultrapure water (UPW) was obtained from Elga Purelab Ultra purification system. QuEChERS salts (6 g anhydrous MgSO4 and 1.5 g NaCl) and dispersive-solid phase extraction (SPE) sorbents in 15-mL polypropylene (PP) centrifuge tubes (900 mg MgSO4, 300 mg PSA, 150 mg graphitized carbon black [GCB]) were supplied by UCT. PP centrifuge tubes were from Sarstedt. Disposable PP syringes were obtained from Becton Dickinson. Nylon syringe filters (15 mm, 0.2 μm), and PP vials (1 mL) with caps were from Agilent Technologies.


Method validation was performed with the use of whole milk, RTF and powdered infant formula, milk powder, soybean oil, and maltodextrin samples obtained from local retail market and previously demonstrated to be free of target PFAS.
Sample Preparation
Sample preparation followed a modified QuEChERS method based on the U.S. FDA method C-010.01.
Homogenous samples (2 g for solids and oils and 5 g for liquids) were weighed into a 50-mL polypropylene (PP) tube followed by the addition of UPW (15 mL for solids and oils and 5 mL for liquids). Hexane (10 mL) was added to oil samples prior to aliquoting UPW. Each sample was spiked with 10 ng of isotope labeled PFAS, added with 10 mL acetonitrile and 150 μL formic acid. The tube was vortexed for 1 min. Preweighed QuEChERS salt mixture was added to the tube, and the mixture was immediately shaken to ensure crystalline agglomerates were broken up sufficiently. Samples were further vortexed on a multi-tube vortexer set to 1500 rpm for 5 min. Following centrifugation at >2,000 x g, the upper acetonitrile layer was transferred into a 15-mL tube while avoiding lipid layer or hexane. In the next step, preweighed dispersive-SPE sorbent mixture (900 mg MgSO4, 300 mg PSA and 150 mg GCB) was added to the sample extract, and the tube was vortexed for 2 min and centrifuged (>2,000 x g, 5 min). A 5 mL aliquot of the supernatant was transferred into a fresh 15-mL centrifuge tube, and evaporated at 60 °C to near dryness using a gentle stream of nitrogen. The residues were reconstituted in 500 μL of methanol, and briefly vortexed to mix. The sample was filtered through a 0.2 μm nylon filter into a PP autosampler vial, and capped.
LC–MS/MS Instrumentation
LC–MS/MS analysis of the PFAS was carried out using an Agilent 1290 Infinity II liquid chromatograph (LC), coupled with an Agilent 6495 triple quadrupole LC–MS system.
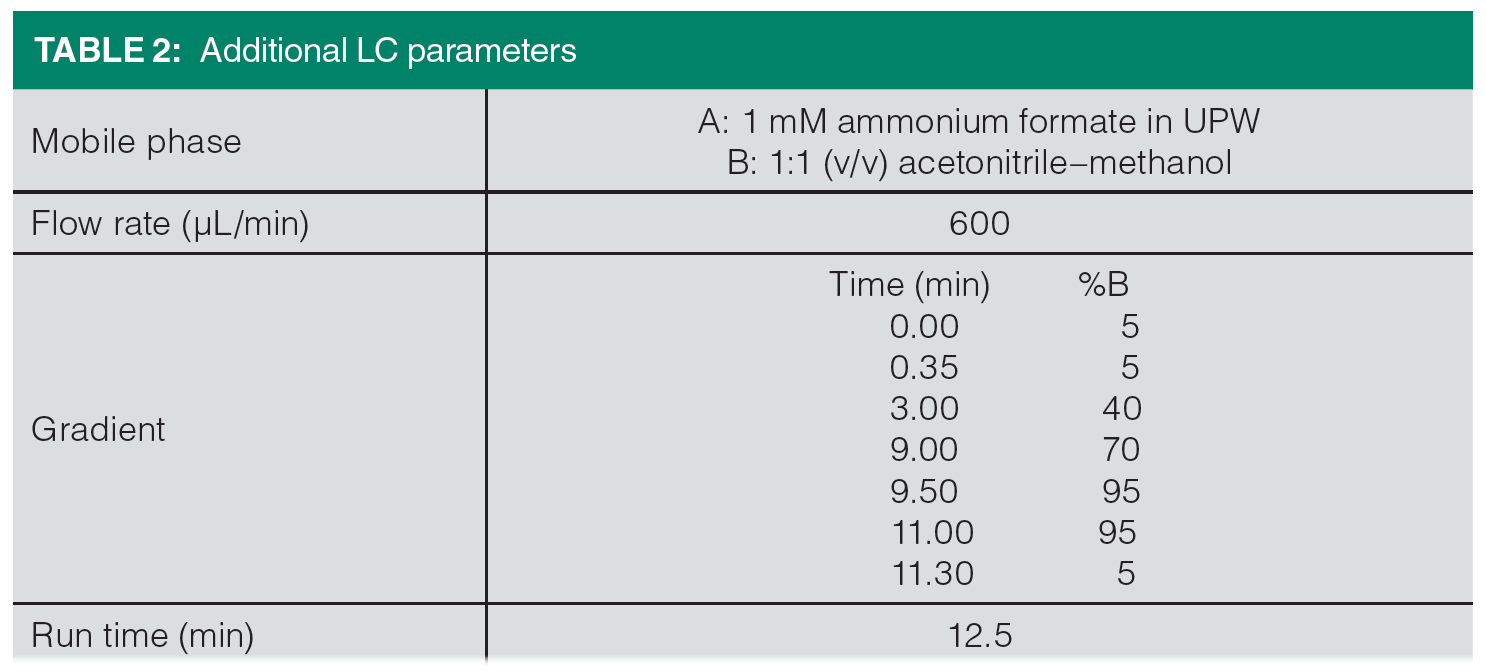
The LC parameters are listed in Table 2. The 1290 Infinity II LC was equipped with an Agilent Zorbax Eclipse Plus rapid resolution high definition (RRHD) C18 column (2.1 x 50 mm, 1.8 μm) with an Agilent Zorbax Eclipse Plus guard column. Gradient elution was performed with 1 mM ammonium formate in UPW (A) and a mixture of acetonitrile and methanol (1:1, v/v) (B) at a flow of 600 μL/min. The eluent was diverted to waste during the first 1.0 min and after 10 min of the run. The total run time from injection to injection was approximately 12.5 min.

To eliminate PFAS background, an Agilent perfluorinated compound (PFC)-free high-performance liquid chromatography (HPLC) conversion kit was installed on the Agilent 1290 Infinity II LC system (5). Additionally, an Agilent Poroshell 120, 3.0 x 30 mm, 1.9 μm delay column was connected between the solvent mixer and injector module to further control potential background contamination from the mobile phases. To reduce contamination due to sorption after injection, the needle wash procedure consisted of 10-sec washes with acetonitrile:isopropanol (1:1, v/v), followed by methanol and UPW:methanol (9:1, v/v).
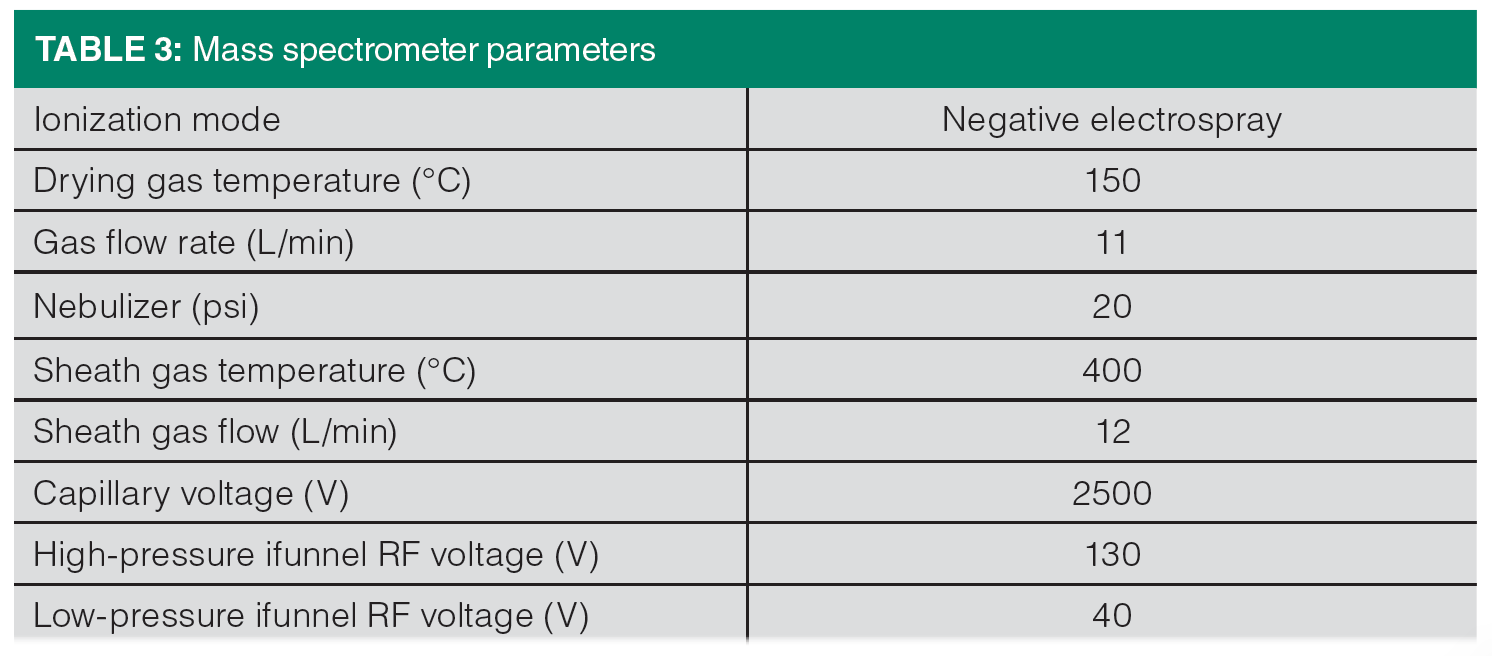
The LC–MS parameters are listed in Table 3. The mass spectrometer was equipped with an electrospray ionization (ESI) source and operated in negative ion and multiple reaction monitoring (MRM) modes. The MRM parameters (listed in Table 1) were optimized for best response using the Agilent MassHunter Optimizer software.

Quantification of target analytes was performed with the use of calibration standards prepared in solvent. Isotope-labeled internal standards were employed to normalize analyte response. Analyte identification in samples was accomplished via retention time and qualifier-to-quantifier MRM ratio comparison with calibration standards.
Results and Discussion
Method Optimization
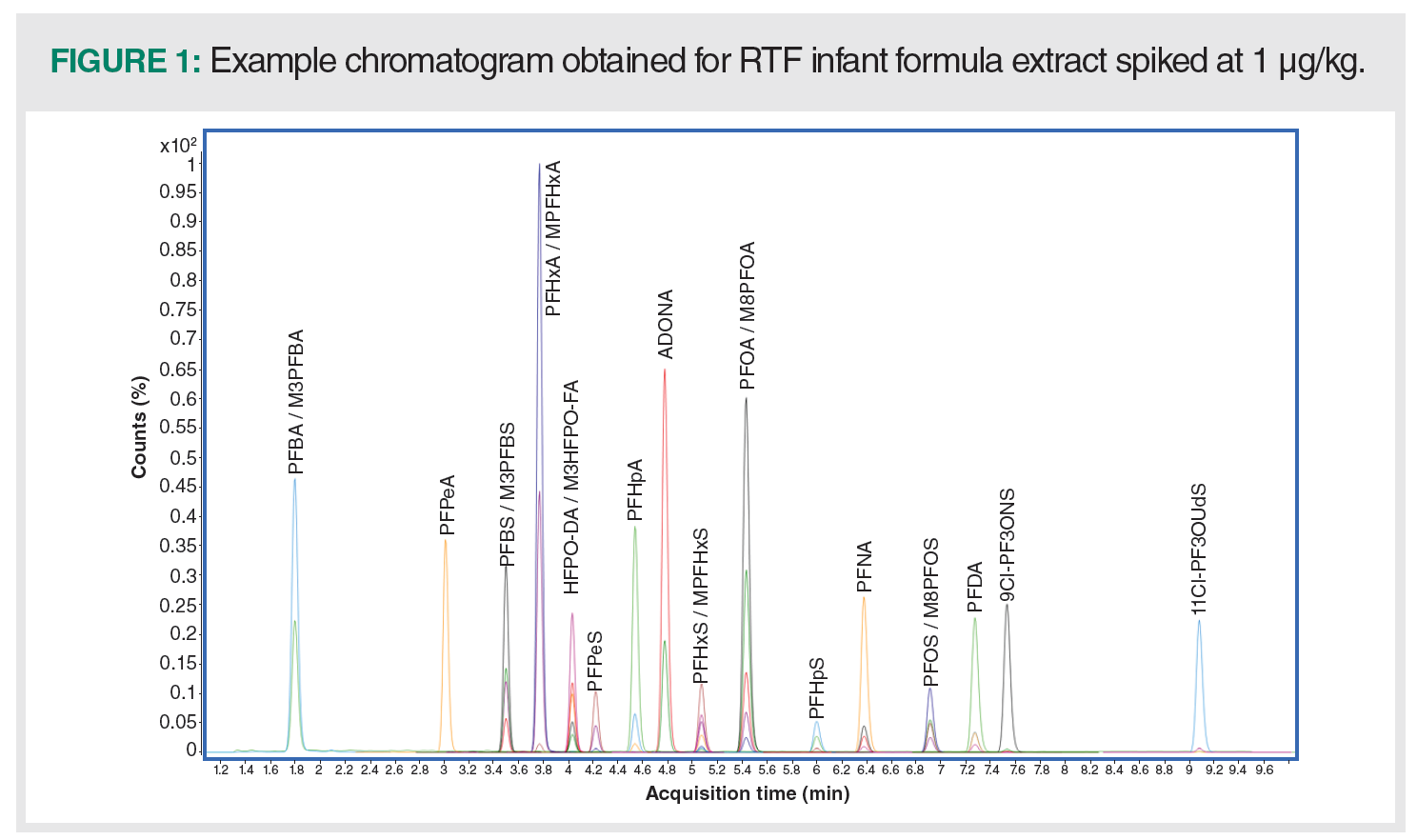
The main focus of the optimization efforts was on high throughput of the analysis. To meet this requirement, a 50 mm ultrahigh-pressure liquid chromatography (UHPLC) C18 column with sub-2-micron particle size and a corresponding 5 mm guard column maintained at 50 oC was used. Under the optimized conditions, baseline separation of all analytes was achieved in approximately 9 min, a significant improvement over the U.S. FDA method C-010.0 (4). The analyte peak widths at the baseline were below 0.2 min. A representative chromatogram of RTF infant formula extract spiked with target analytes at a concentration of 1 μg/kg is presented in Figure 1.

Target precursor ions represented deprotonated molecules for all analytes except for hexafluoropropylene oxide-dimer acid (HFPO-DA, or Gen X) that fragmented during the ionization to form a decarboxylation product. Considerable time was spent evaluating the type and concentration of modifier in the aqueous mobile phase component A. Various concentrations of ammonium acetate, ammonium fluoride, and ammonium formate were tested. There was only minimal impact on separation, but the type and concentration of the modifier impacted the response of certain analytes, especially HFPO-DA. Using 1 mM ammonium formate as mobile phase A allowed for the best response for HFPO-DA, while not sacrificing response for the other PFAS.
Method Validation
The method validation evaluated selectivity, linearity, accuracy and precision, limit of quantification (LOQ), and method detection limit (MDL).
Selectivity of the method was demonstrated based on analyses of unspiked samples and a method (procedural) blank in each validation batch. The responses of target PFAS in matrix and method blank injections were <30% of the response in samples spiked at the LOQ. Analytes present in validation samples at or above the LOQ were successfully identified. The analyte retention times in samples were well within 0.1 min of the retention time in standards. Ion ratio in samples were within ±30% of the average ratio established in calibration standards for at least one quantifier/qualifier transition pair. Ion ratio-based identification could not be performed for perfluorobutyrate (PFBA) and perfluoro-n-pentanoic acid (PFPeA), since only a single MRM transition was available for each of these analytes.
Linearity was evaluated with each validation batch at eight concentration levels ranging from 0.1 to 50.0 ng/mL injected at minimum at the beginning and at the end of the measurement sequence. This concentration range corresponded to 0.050 to 25 μg/kg and 0.020 to 10 μg/kg in 2-gram and 5-gram samples, respectively. Linear regression calibration curves with 1/x weighting were constructed by plotting the analyte-to-internal standard peak area ratio against the analyte concentrations. The coefficients of determination (R2) were consistently above 0.995 for all analytes with residuals within ± 20%.
Accuracy (marginal recovery) and precision [repeatability as relative standard deviation (RSDr) and intermediate precision (RSDINT)] were determined based on replicate analyses of unspiked samples and samples spiked with target PFAS at multiple concentrations ranging from 0.05 to 5.0 μg/kg. The spiking experiments were performed at minimum in six replicates over two different days. The mean recoveries calculated at individual spiking levels above method LOQ were within 70–120% with RSDr and RSDINT values ≤20% indicating robust recoveries in a variety of matrices with high reproducibility. Mean recoveries obtained at LOQ for each analyte are shown in Figure 2.

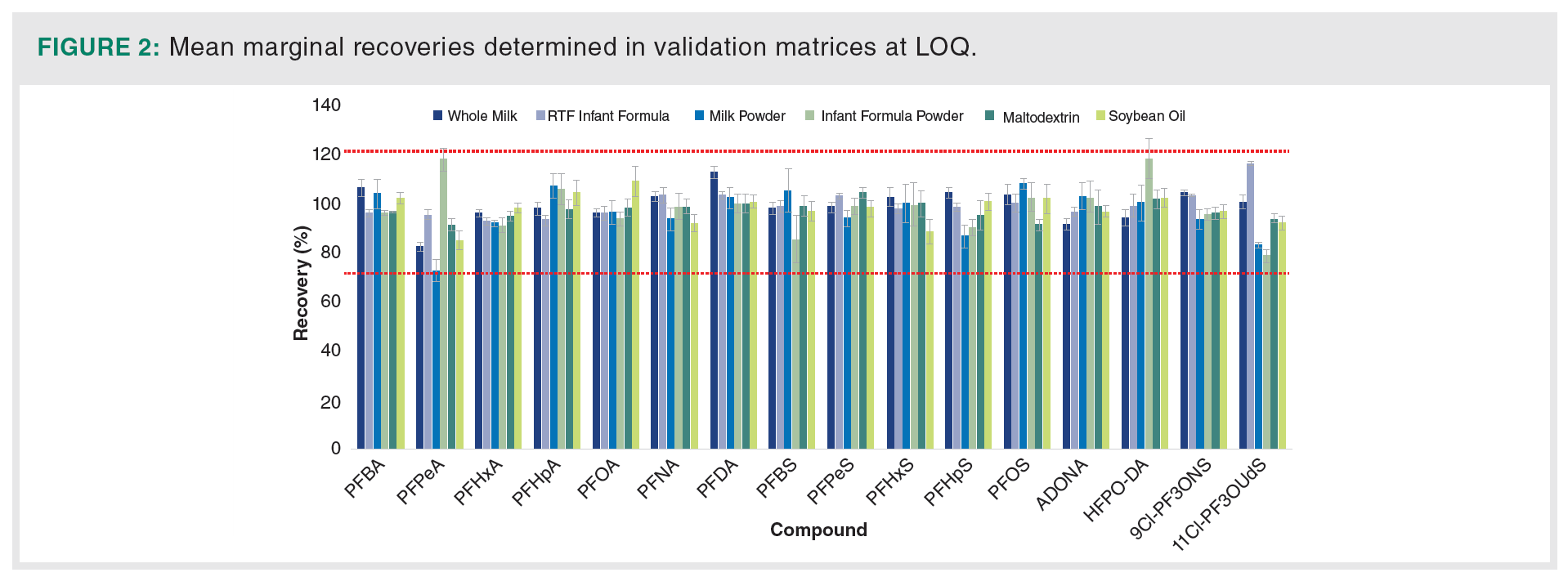
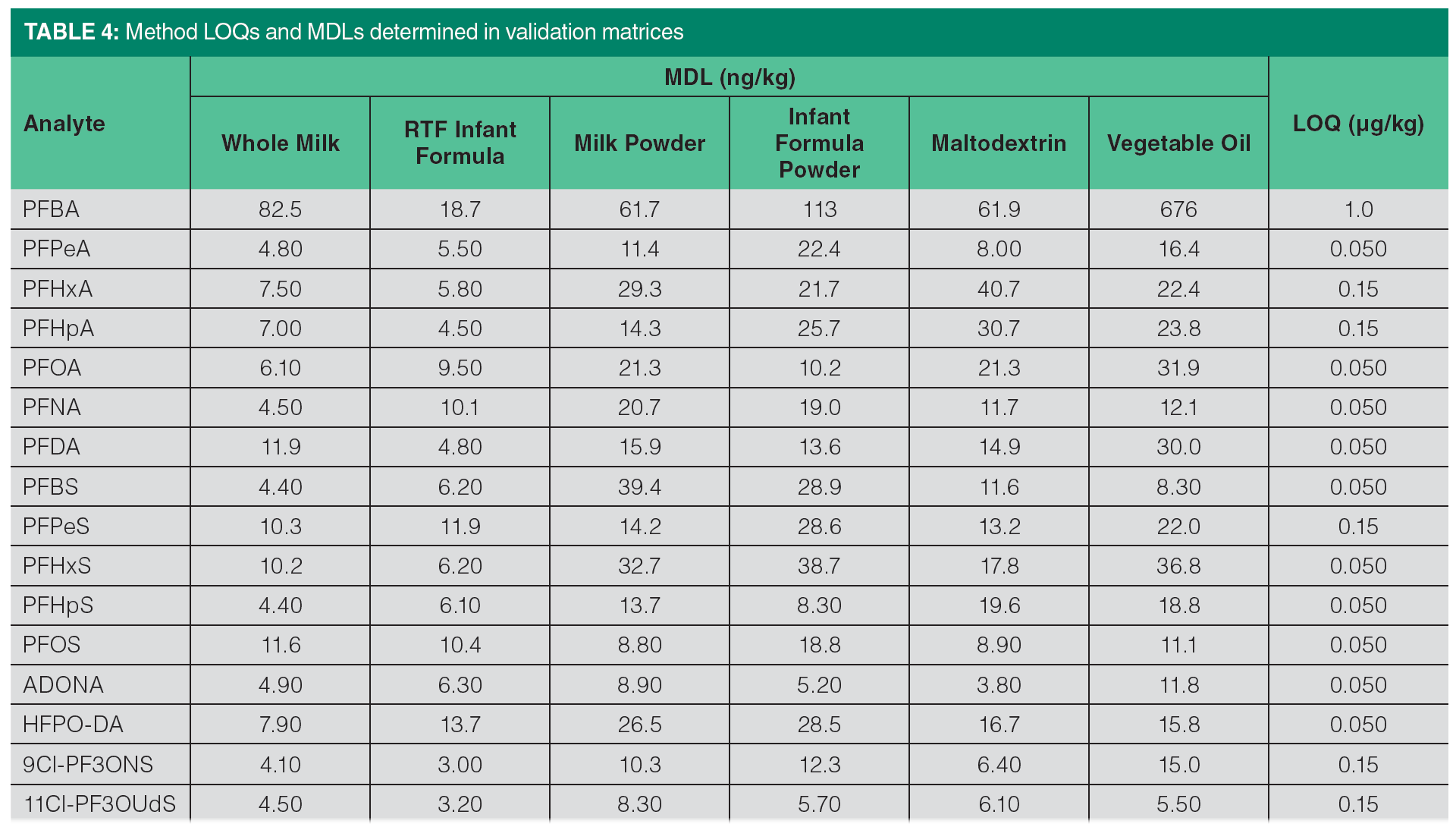
Method detection limit (MDL) was determined for each analyte/matrix combination based on accuracy and precision data after samples were taken through the entire workflow including sample extraction and LC–MS/MS analysis. Standard deviation was calculated based on precision data at low level spiking concentration and the obtained value was multiplied by t-value corresponding to the appropriate degrees of freedom (6). LOQ was determined for each PFAS as the lowest spiking level for which the recoveries within 70–120 and RSDs ≤20% were achieved in all matrices while successfully identifying the analyte. MDLs and LOQs are summarized in Table 4. MDLs were typically lower than 25 ng/kg for most PFAS analytes in all matrices except for PFBA which was significantly higher in matrices other than infant powder.

Conclusions
This paper described optimization and validation of a sensitive, robust and high-throughput LC–MS method based on modified QuEChERS procedure for the determination of sixteen priority U.S. FDA PFAS analytes including perfluoroalkyl carboxylic acids, perfluoroalkyl sulfonic acids and major replacement chemicals in whole milk, infant formula and related ingredients. The validation experiments demonstrated adequate performance of the method in the studied matrices that proves to be robust, reliable and reproducible with scope to expand the list of PFAS in the future.
References
- B.J.A. Berendsen et al., Food Addit. Contam. A 37, 1707–1718 (2020).
- “EFSA Scientific Opinion: Risk to Human Health Related to the Presence of Perfluoroalkyl Substances in Food.” EFSA Journal 18(9), 6223 (2020). DOI: https://doi.org/10.2903/j.efsa.2020.6223
- M.M. Al Amin et al., Environ. Technol. Innov. 19, 100879 (2020). DOI: https://doi.org/10.1016/j.eti.2020.100879
- Method C-010.01: Determination of 16 Perfluoroalkyl and Polyfluoroalkyl Substances (PFAS) in Food using Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/ MS), FDA Foods Program Compendium of Analytical Laboratory Methods: Chemical Analytical Manual (CAM).
- M. Kamuf et al. “Reduce PFAS Background with the Agilent PFC-Free HPLC Conversion Kit.” Agilent Technologies Application Note, p/n 5994-2291EN (2020).
- U.S. Environmental Protection Agency, “Definition and Procedure for the Determination of the Method Detection Limit” 40 CFR 136, Appendix B, revision 2 (2016).
About The Authors

Lukas Vaclavik (above), John Schmitz, Matthew Eckert, and Katerina Mastovska are with Eurofins Food Chemistry Testing, in Madison, Wisconsin. Tarun Anumol is with Agilent Technologies, Inc., in Little Falls, Delaware. Direct correspondence to: LukasVaclavik@eurofins.co.uk











New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.


Analytical Challenges in Measuring Migration from Food Contact Materials
November 2nd 2015Food contact materials contain low molecular weight additives and processing aids which can migrate into foods leading to trace levels of contamination. Food safety is ensured through regulations, comprising compositional controls and migration limits, which present a significant analytical challenge to the food industry to ensure compliance and demonstrate due diligence. Of the various analytical approaches, LC-MS/MS has proved to be an essential tool in monitoring migration of target compounds into foods, and more sophisticated approaches such as LC-high resolution MS (Orbitrap) are being increasingly used for untargeted analysis to monitor non-intentionally added substances. This podcast will provide an overview to this area, illustrated with various applications showing current approaches being employed.
 Headquarters complex in Washington, DC. | Image Credit: © Tada Images - stock.adobe.com")




