Welcome to the Brave New World of CSV?


Data integrity issues are changing the way that we should be undertaking computerized system validation (CSV) of our chromatography data systems (CDSs). Do you understand what is required in the brave new world of CSV?
R.D. McDowall, R.D. McDowall Ltd, Bromley, Kent, UK
Data integrity issues are changing the way that we should be undertaking computerized system validation (CSV) of our chromatography data systems (CDSs). Do you understand what is required in the brave new world of CSV?
“Our CDS is validated” is a common statement I hear when training or consulting. This column has discussed different aspects of the validation of chromatography data systems (CDSs) for many years and has featured some case-study examples of validation from quality control or bioanalytical laboratories (1,2). The aim of these Questions of Quality columns is to help readers understand that computerized system validation (CSV) is not rocket science or brain surgery, but the application of good software engineering practice principles in the context of a regulated chromatography laboratory. CSV is not a typical skill for a chromatographer but the principles are not difficult to comprehend and can be easily understood over the course of a validation project. However, the CSV world is changing - let us see how.
The Way it is Now
Traditionally CSV in a regulated context uses a rather old-fashioned life cycle V model to explain how to perform a CDS validation; this is presented in Figure 1 and has been adapted for a laboratory system such as a CDS. In overview, the validation plan and validation summary report are the controlling documents that define the work life cycle phases to be undertaken and report what was actually performed. In more detail the validation plan will define the tasks to be performed in each phase together with the documented evidence required to support the claim that the system is validated. The people involved with the validation are listed along with their responsibilities. The report should mirror the plan and describe the actual work performed plus explain any differences from the validation plan.


Figure 1: Typical life cycle model for a chromatography data system.
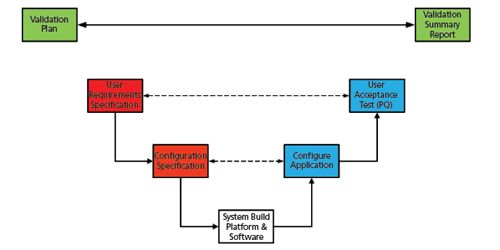
On the left-hand side of the figure, the specification of the system is contained in a user requirements specification (URS), in addition to how the CDS application will be configured in a document strangely called the configuration specification. Together the two documents define the intended purpose of the system as required by the regulations (3,4). The underlying computer platform and operating system, followed by the installation of the various components of the CDS are installed, qualified, and integrated into a basic unconfigured system shown at the bottom of the V in Figure 1. Next, the CDS software is configured as defined in the configuration specification; for example, by turning on or off functions in the software to change the business process to match the laboratory requirements; the use of electronic signatures; defining the user types and the corresponding access privileges; functions to protect electronic records, etc. Finally, the configured CDS is tested against the requirements in the URS. As shown, there is symmetry of the V model with an activity on the left-hand side that is matched by a corresponding activity on the right. This is similar to a chemical reaction - validation does not work unless the two sides of the equation (or V) are balanced.
And now, hey presto, the system is validated! Or it would be if you have performed tasks like process redesign; traceability of requirements; writing procedures to use the system; writing procedural controls to plug regulatory compliance gaps (workarounds); IT support agreements; training users; implementing custom calculations; and designing custom reports; however, the bulk of the work is outlined in Figure 1(5).
What a V model does not describe is how the application should be introduced into a laboratory.
Process, Process, Process
One of the items not covered in Figure 1 is the understanding of the chromatographic process and how it can be redesigned using the introduction of a new version of an existing CDS or a new CDS to make the process more efficient. Typically, this would be done so that electronic signatures and electronic working can be used, with the elimination (or perhaps extermination would be a better word) of all those horrible spreadsheets that slow down the process. It never ceases to amaze me why chromatographers are so stupid in this respect. An organization spends millions on a shiny CDS that is capable of amazing things only for idiots in the laboratory to print out piles of paper and then enter data manually into a spreadsheet and carefully check the entries. Perhaps if Dante were to rewrite his Inferno and set it in modern times, this would be his vision of chromatographic hell. Endless manual data entry and transcription error checks performed forever in an ocean of paper. This would be coupled with the devils from Hell’s QA department poking those miscreants who did not spot a transcription error with sharpened poles. Perhaps this is a description of your laboratory?
In an ideal world we would be working electronically. The way that this would be achieved is to redesign the process, as shown in Figure 2. The CDS application would then be configured to match the resigned process. The focus is on a top-down approach aimed at the process efficiency.

Figure 2: Traditional computer validation focuses on the process.
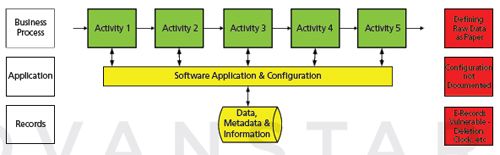
The validation of a CDS therefore couples the life cycle tasks outlined in Figure 1 with the process redesign shown in Figure 2. Now the system is validated! We can now relax safe in the knowledge that things are under control.
Data Integrity and Potential Problems
The above has been the way most validation work has been performed. However, there are three potential problems that may arise in this approach (Figure 2):
1. Process level: A problem arises if the system is used as a hybrid and paper is defined as the raw data. Oh dear! The FDA shot this argument down in flames in 2010 with a Level 2 guidance where they stated that paper was neither a true copy nor an exact and complete copy of the underlying electronic records (6). This column has discussed this in detail when looking at complete data for a CDS (7,8).
2. Application level: The CDS configuration settings are not documented or the settings do not protect the electronic records, for example, the audit trail functions have not been enabled. This is unwise because inspectors have been trained to request this documentation. Hence, one should understand the ramifications contained in Figure 1.
3. Record level: Protection of electronic records created and managed by the application. We will discuss this issue in more detail later. However, if your electronic records are stored in directories in the operating system - be afraid, be very afraid.
Beneath the application in Figure 2 are the data and metadata produced from the analyses performed in the laboratory. For a more detailed discussion of the records that constitute a primary analytical record see the last Questions of Quality column by Burgess and McDowall (9). Let us look at these records in more detail.
With a CDS there are two options for storing the data: either in directories in the operating system file structure or in a database. McDowall and Burgess recently published a trilogy of papers in four parts in LCGC North America that looks at the ideal chromatography data system for a regulated laboratory (10–13), in the paper on system architecture we recommended that standalone workstations are not fit for purpose and that a CDS must store the data and contextual metadata in a database (11). Records stored in directories are too vulnerable to deletion and unrestricted access to the system clock can enable time travelling on a standalone workstation. A user can access the system clock and put the clock back in time, delete failed records, repeat the work, pass the batch, and no-one is the wiser! To be secure, data must be stored on a fault tolerant network drive where the clock source is a time server linked to a trusted time source, with effective and regular backup performed by the IT department.
Please note that data integrity is not a simple, single discipline issue solely in the chromatographic domain. Rather it is a multi-disciplinary function that requires a mix of people who have between them IT, regulatory compliance, software engineering, and business knowledge skills. People with cross-disciplinary skills are invaluable here (14).
Validated System with Vulnerable Records?!
Let us consider the following situation: We have a CDS (standalone or networked) where the electronic records generated by the system are stored in directories within the operating system. If we have validated the system taking the approaches outlined in Figure 1 and Figure 2, there is still a possibility that the records can be inadvertently deleted or manipulated. Where does our validation stand now? Application under control but records potentially vulnerable? Not the best situation to be in, is it? What should we do - apart from panic or ensure our CVs are current?
Note that this discussion is CDS specific, but the principles outlined here are also applicable to other standalone laboratory systems and PC instrument controllers.
Back to the Future?
To go forward let us go back in time approximately 10 years. In 2005 the GAMP Forum (Good Automated Manufacturing Practice) published a Good Practice Guide (GPG) on Compliant Part 11 Electronic Records and Signatures (15). The approach was rather different to the way I have described validation above. Instead of the top-down validation approach, they took a bottom-up approach and focused on the electronic records and signatures created and used within the system. In overview, the process was to identify the records created in the system, evaluate their regulatory impact, and, as a result, determine the controls that were necessary to control and protect them. What happened? The validation world listened with deaf ears and saw with blind eyes.
I believe the problem is that this approach does not create process efficiencies that the top-down approach does; a focus on records creates protected records but you can still have an inefficient process. However, it is time to reconsider the bottom-up approach.
Brave New CSV World?

Figure 3: Computerized system validation using a combination of top-down and bottom-up approaches.
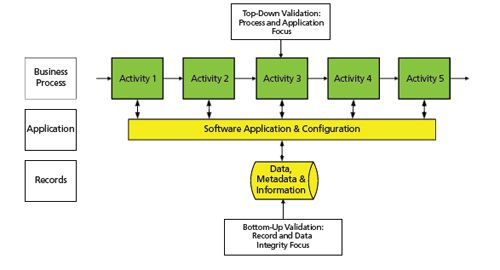
I would suggest a hybrid of both approaches to get the best of both worlds and to ensure the integrity of our electronic records. With little additional effort but with great compliance benefit, the vulnerability of the electronic records should be managed by controls specifically implemented, which are based on the record’s regulatory impact. This is shown in Figure 3 and would proceed in a number of stages:
1. The start of the project would be a focus on process improvement and efficiency gains.
2. As the selected application was prototyped and configuration settings of the CDS examined, all applicable electronic records generated in the course of analysis (data and metadata including audit trail entries) would be identified.
3. The regulatory impact of the records would be assessed depending on their function; for example, method development, method validation batch release or protocol analysis, stability testing, etc.
4. The vulnerability of the electronic records would be assessed and appropriate controls to protect these records would be added to the specification documents for implementation in the later stages of the validation project.
5. As the system is being built, controls for the electronic records and signatures would be implemented at the same time as application configuration. These controls can be either technical or procedural.
6. During the performance qualification (PQ) or user acceptance testing (UAT) phase of the validation the additional controls for the records and signature would be integrated into the overall testing of the intended use of the CDS application.
Turning Principles into Practice
That may be the principles but you may be thinking that a few fancy diagrams do not give sufficient detail to the approach. Let us take the principles above and turn them into practice here. We join the validation of a new CDS at the prototyping phase where the application is being configured and the Part 11 controls are being evaluated. The CDS is being installed in a regulated Quality Control laboratory undertaking verification of compendial methods, and analysis of active ingredients, in-process materials, and finished goods. Stability testing is also performed. The project team decide that electronic signatures and the 21 CFR 11 controls offered by the application will be implemented. Although the application is networked, all data are stored in directories in the operating system and not in a database. Snatching defeat from the jaws of victory for the selection team!

Figure 4: Identification of controls for high, medium, and low Impact regulatory records (see reference 16).
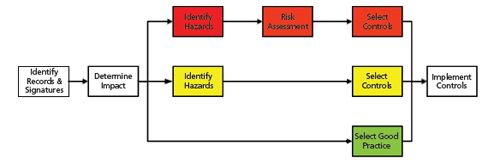
The process for bottom-up or records-based validation is outlined in Figure 4 and each stage is described below:.
The first task is to identify the electronic records and signatures generated and maintained in the system (9).


Next the regulatory impact of the identified records or signatures needs to be assessed. The GAMP Part 11 GPG classifies records into high, medium, and low impact categories as shown in Table 1. From the descriptions of the use of the system and the table, the CDS records fall into the high impact category because they are involved in product release.
The identification of any hazards that the records face is now performed followed by a risk assessment (all documented). To expedite the process, we will assume that this has been done. At the highest risk are the records on the server hard drive in the operating system directories because they can be deleted outside of the application, without leaving any evidence of their deletion.
Controls need to be selected to protect these high risk records; for example, records can only be accessed by authorized users via the application, restricting access to directories by a shell programme, hiding the drive on the network, monitoring access to the drive via the operating system, restricting copy of CDS records. These controls need to be documented in the specification(s) for the CDS.
As the validation progresses the controls will be implemented and later tested as part of the user acceptance tests for the system.
Summary
This short example gives you a better idea to of how to ensure that both a top-down and bottom-up approach to validation, as shown in Figure 3, provides business benefits while at the same time implements controls that will help ensure data integrity. Data integrity is not just the domain of the laboratory but requires a multi-disciplinary team to assess record vulnerability and to incorporate the controls within a CDS validation.
Acknowledgement
I would like to thank Mark Newton for his comments in preparing this column.
References
- J. Donath and R.D. McDowall, LCGC Europe18(9), 453–464 (2005).
- T.D. Thompson, D. Browne, D. Mole, and R.D. McDowall, LCGC Europe14(11) 687–692 (2001).
- Current Good Manufacturing Practice for Finished Pharmaceutical Products, 21 CFR 211.63 (2008).
- EU GMP Annex 11 computerized systems (2011).
- R.D. McDowall, Spectroscopy 21(4), (2006).
- Questions and Answers on Current Good Manufacturing Practices, Good Guidance Practices, Level 2 Guidance - Records and Reports
- http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm124787.htm
- R.D. McDowall, LCGC Europe26(6), 338–343 (2013).
- R.D. McDowall, LCGC Europe 26(7), 389–392 (2013).
- C. Burgess and R.D. McDowall, LCGC Europe28(11), 621–625 (2015).
- R.D. McDowall and C. Burgess, LCGC North America33(8), 554–557 (2015).
- R.D. McDowall and C. Burgess, LCGC North America33(10), 782–785 (2015).
- R.D. McDowall and C. Burgess, LCGC North America33(12), 914–917 (2015).
- R.D. McDowall and C. Burgess, LCGC North America Scheduled February 2016.
- Mark E. Newton, personal communication.
- Good Automated Manufacturing Practice (GAMP) Good Practice Guide, Compliant Part 11 Electronic Records and Signatures, ISPE, Tampa, Florida, USA, (2005).
“Questions of Quality” editor Bob McDowall is Director at R.D. McDowall Ltd, Bromley, Kent, UK. He is also a member of LCGC Europe’s editorial advisory board. Direct correspondence about this column should be addressed to the editor-in-chief, Alasdair Matheson, at amatheson@advanstar.com









What Goes in a CDS IT Service Level Agreement?
Published: April 7th 2025 | Updated: April 7th 2025Protecting your network chromatography data system (CDS) data is critical and a service level agreement (SLA) with your IT provider is vital. What should be included? Are SLAs for in-house IT and SaaS (software as a service) similar?






