The Rise of Hydrophilic Interaction Chromatography in Untargeted Clinical Metabolomics
LCGC Europe
Hydrophilic interaction chromatography (HILIC) was introduced more than two decades ago and has garnered much attention. Characterized by a hydrophilic stationary phase used in combination with an aqueous organic mobile phase, numerous improvements have been achieved and HILIC is now considered as an attractive alternative to reversed-phase phase liquid chromatography (LC) for many applications. HILIC provides several advantages over reversed-phase LC for the analysis of polar compounds, including higher retention of polar metabolites, enhanced mass spectrometric sensitivity, moderate back-pressure - even at high flow rates, or when used with sub-2-µm particle size - and orthogonal selectivity. Several important technical developments have been proposed during the last decade that foster its use in metabolomics. This review presents an overview of the most recent technical improvements and applications of HILIC analysis in untargeted clinical metabolomics and discusses important practical considerations, including the selection of the optimal column chemistry, appropriate eluents, sample preparation, and data analysis.
Isabelle Kohler, Rico J.E. Derks, and Martin Giera, Center for Proteomics and Metabolomics, Leiden University Medical Center, Leiden, the Netherlands.


Photo Credit: Witchaphon Saengaram/Getty Images

Hydrophilic interaction chromatography (HILIC) was introduced more than two decades ago and has garnered much attention. Characterized by a hydrophilic stationary phase used in combination with an aqueous organic mobile phase, numerous improvements have been achieved and HILIC is now considered as an attractive alternative to reversed-phase phase liquid chromatography (LC) for many applications. HILIC provides several advantages over reversed-phase LC for the analysis of polar compounds, including higher retention of polar metabolites, enhanced mass spectrometric sensitivity, moderate back-pressure - even at high flow rates, or when used with sub-2-µm particle size - and orthogonal selectivity. Several important technical developments have been proposed during the last decade that foster its use in metabolomics. This review presents an overview of the most recent technical improvements and applications of HILIC analysis in untargeted clinical metabolomics and discusses important practical considerations, including the selection of the optimal column chemistry, appropriate eluents, sample preparation, and data analysis.
Metabolomics refers to the analysis and the study of the metabolome which encompasses the totality of the metabolites of a biological system, that is, the intermediate and end-point products of metabolism (1). Together with other omics technologies, including genomics and pharmacogenomics, metabolomics is currently recognized as the core of personalized medicine, which aims to predict the individual susceptibility to a disease or outcome before a therapeutic intervention (2–4).
The practical value of metabolomics, which is used as a tool to investigate metabolic phenotypes linked to genetics and environmental effects, has already been demonstrated for a number of diseases including metabolic syndrome, diabetes, and infections (5–7). While early metabolomics studies focused on the discovery of single molecules as specific biomarkers, it is now widely accepted that pathological processes rely on much more complex metabolic changes, which places metabolomics at the core of dissecting pathological phenotypes (8).
As well as the large number of metabolites that can be present in biological fluids, their physico-chemical diversity and wide range of concentrations (often several orders of magnitude) represents a major analytical challenge and no single analytical platform is currently able to provide a complete coverage of the metabolome. Nuclear magnetic resonance (NMR) and mass spectrometry (MS) based techniques, including liquid chromatography (LC), gas chromatography (GC), and capillary electrophoresis (CE) are commonly used in metabolomics, often in combination. LC coupled to MS, especially in reversed-phase chromatographic mode, remains the gold standard technique for untargeted metabolomics. Reversed-phase LC is characterized by high sensitivity, separation efficiency, and throughput, particularly when used in combination with sub-2-µm particle size columns (ultrahigh-pressure LC [UHPLC]) or superficially porous particles (core–shell or fused-core technology). Reversed-phase LC is suitable for the analysis of a large fraction of biological compounds but highly polar molecules such as saccharides, phosphates, amino acids, nucleic acids, and short-chain organic acids are poorly retained on typical reversed phase stationary phases (9,10). This is a clear limitation because they represent a large fraction of important endogenous metabolites (11). The poor retention behaviour of polar metabolites under most reversed phase conditions also leads to co-elution with matrix interferences, resulting in significant matrix effects during electrospray ionization (ESI), and leading to poor sensitivity and inaccurate data (12). Several attempts have been proposed to increase the retention of polar compounds in reversed-phase LC, for example, by using polar-embedded or polar-endcapped groups in combination with C18 phases; adding ion-pairing agents to the mobile phase for charged compounds; or derivatizing the polar functional group into a more hydrophobic one (13,14). However, all of these strategies suffer from limitations in terms of instrument fouling, sensitivity, or partial metabolite coverage. Hydrophilic interaction chromatography (HILIC) is therefore considered as an effective approach to analyze the polar fraction of the metabolome.
In HILIC, a term first coined by Alpert in 1990 (15), a polar stationary phase is used with a relatively hydrophobic mobile phase composed of an aqueous-organic mixture usually containing approximately 5% to 40% of water, forming a water-enriched layer of partially immobilized eluent on the stationary phase. The stationary phase generally consists of underivatized bare silica or silica modified by functional groups such as amides, diols, amines, or zwitterionic moieties. The mobile phase is composed of water-miscible aprotic polar organic solvents, such as acetonitrile, used with volatile buffers (16). The retention mechanisms occurring in HILIC are rather complex and not yet completely understood but involve hydrophilic partitioning, dipole–dipole interaction, hydrogen bond, and electrostatic interaction (11,13,16–19). HILIC shows several advantages compared to reversed-phase LC, such as an increased analyte diffusivity in the organic-rich mobile phase (lower C-term contribution to the Van Deemter curve) (20); lower backpressure because of low viscosity of the mobile phase (21); enhanced MS signal as a result of better eluent desolvation (increased ionization efficiency) (22,23); and the possibility of direct injection of organic extracts as usually obtained by protein precipitation or extraction (16,24).
Even though multiple studies over the last decade have demonstrated the usefulness of HILIC, it remains rarely used compared with reversed-phase LC in untargeted metabolomics. This reluctance may be because HILIC is less flexible and not as straightforward as reversed-phase LC. Indeed, because of the complexity of the retention mechanisms, certain practical aspects should be considered to ensure high quality data. The following sections provide a practical guide for the implementation of HILIC in untargeted clinical metabolomics.
Sample Preparation
When using HILIC, an optimal sample preparation is not only critical for ensuring maximal recovery of a large range of metabolites but also for chromatographic separation performance. Untargeted metabolomics implies the analysis of the highest number of metabolites and, hence, a sample pre-treatment as simple as possible. Human and animal samples used for clinical metabolic profiling include a wide range of biological matrices such as blood-derived samples, urine, saliva, tears, cerebrospinal fluid, tissues, and faeces (25). Among them, blood plasma and urine represent by far the majority of applications and their pre-treatment is discussed in this article.
Plasma Samples: Plasma is usually preferred over serum because of artifact formation, which may occur during blood clotting (26,27). However, plasma has an overall higher protein content, underlining the importance of an efficient protein removal step. Protein precipitation (PP) by the addition of cold organic solvent is the most commonly used preparation method because it allows for a simultaneous quenching and non-selective sample pre-treatment. Moreover, solvent-based PP techniques lead to higher metabolite coverage compared to heat or organic acid treatment (28). The composition and volume of the organic solvent has a major effect on both the metabolic coverage and the protein removal. In general, acetonitrile and acetone give better results in terms of protein removal efficiency while methanol and ethanol (EtOH), usually in a ratio ≥ 2.5:1 (solvent:sample, v/v) result in a higher number of metabolites detected and are commonly considered the first choice when combined with reversed-phase LC analysis (28,29). In HILIC analysis, the nature of the sample diluent has been shown to be much more critical than in reversed-phase LC. It is well known that peak distortion is observed in the presence of large amounts of water in the injected sample, especially for early-eluting peaks because of the significant difference in elution strength between the highly organic mobile phase and the dissolution solvent (30,31). Such effects are also observed when using polar protic solvents such as methanol, EtOH, and isopropanol (iPA). Solvents with higher hydrogen bonding donor capability (water > methanol > EtOH > iPA >> acetonitrile) may disturb the water-enriched layer at the surface of the stationary phase leading to deteriorated peak shapes. It is therefore recommended to use the lowest amount of protic solvents in the sample plug (that is, ≤10–20%, depending on the stationary and mobile phase composition) and inject less than 1% of the column volume (for example, ~2 µL for 100 mm × 2.1 mm i.d. columns). For compounds presenting solubility issues in pure acetonitrile, acceptable peak shapes may be obtained by using a mixture of acetonitrile /iPA (50:50, v/v), provided that the volume injected is reduced to its minimum (31).
Urine Samples: Urine is usually simply filtrated and diluted five- to 10-fold with buffers or water prior to reversed-phase LC–MS analysis (known as dilute-and-shoot). However, in HILIC analysis, this straightforward strategy cannot be applied and urine samples have to be diluted with acetonitrile, leading to precipitation of proteins and salts. Compared to plasma, urine presents a relatively low amount of proteins (0.5–1 g/L). Hence, in high-throughput studies, urine samples are commonly diluted 4–5 times with a mixture of acetonitrile/water 3:1 (v/v). This allows for a sufficient dilution of salts and proteins, which, in turn, enables the direct injection of samples and results in acceptable separation performance. Nevertheless, proteinuria is a common clinical abnormality of many diseases; if samples are suspected to contain a higher amount of proteins, centrifugation and injection of the acetonitrile supernatant is mandatory to avoid on-column precipitation of residual proteins.
While reversed-phase LC is prone to mass overload, HILIC is prone to volume overload phenomena, which may lead to severe peak tailing and distortion, especially if the sample diluent composition is not optimal (24,32,33). Whatever the biological matrix, the injection volume should remain as low as possible, preferentially less than 1% of the total column volume (34). If sensitivity becomes an issue, samples may be pre-concentrated to increase the injected amount while keeping a minimal injected volume.


Chromatographic Separation
Compared with reversed-phase LC, HILIC usually shows lower intra-day and batch-to-batch retention time repeatability, longer equilibration time, and difficulties in predicting the retention behaviour as a result of the multimodal retention mechanism. These limitations do not foster the widespread use of HILIC in large cohort metabolomics studies. However, their impact can be significantly lowered with practical measures, including repeatable preparation of mobile phase (accurate salts concentration and pH titration) as well as sufficient column equilibration.
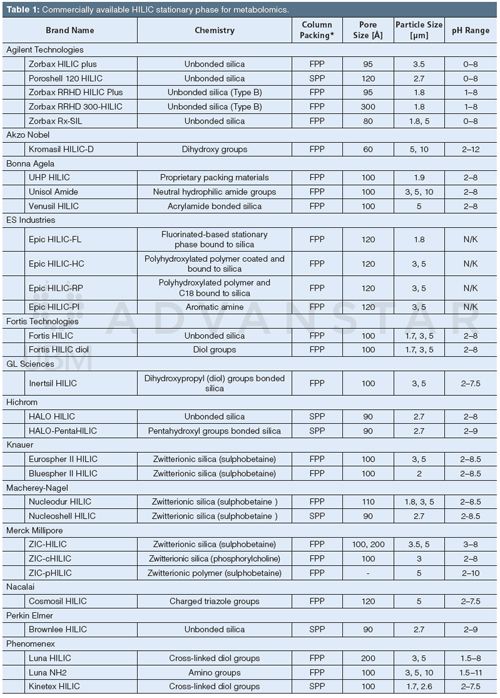
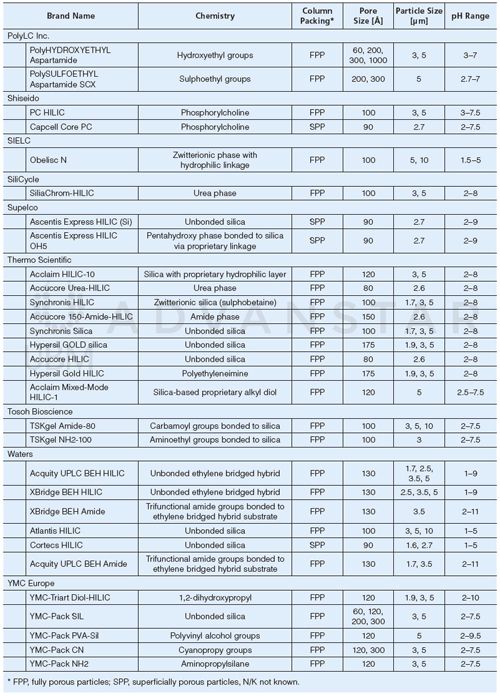
HILIC stationary phases usually consist of bare silica or polymer material modified with polar functional group(s), which allows for hydrophilic partitioning of the analytes between the water-enriched layer and the mobile phase, and other retention mechanisms. As a result of the complexity of HILIC retention mechanism, it is difficult to evaluate the optimal column chemistry based on theoretical assumptions; moreover, the common approaches for reversed-phase LC method development cannot be implemented for HILIC (19,24,35). Hence, HILIC method development, including column selection, is a largely empirical process. Table 1 lists the commercially available HILIC stationary phases (as of November 2015), including the available particle sizes and working pH range. Commonly used chemistries in metabolomics studies include bare silica, diol, and amide, as well as ionic phases such as sulphoalkylbetaine (zwitterionic) and amine, which are of interest in the separation of ionized compounds via additional electrostatic interactions (36). As an example, Figure 1 shows the extracted ion chromatograms (EICs) obtained for the analysis of a set of representative metabolites belonging to different metabolic classes. The differences observed in chromatographic separation depending on the experimental conditions highlight the relative importance of stationary and mobile phases composition. A diol phase (Figure 1[a] and [b]) leads to an acceptable selectivity for the metabolites, which are sufficiently retained while still eluted with a moderate (< 50%) proportion of acetonitrile, leading to shorter analysis times when compared with an amine phase (Figure 1[e]) where acidic compounds are strongly retained as a result of electrostatic interactions. Retention on the cross-linked diol phase has been shown to mostly rely on adsorption and partitioning mechanisms, depending on the proportion of water in the mobile phase (37). Diol phases may lead to poor peak shapes for some compounds with peak tailing or shouldering; hence if peak shape is critical, for example in targeted quantitative metabolomics, the amine phase is an interesting alternative. Amine phases also show an increased peak capacity, particularly for small organic acids, as demonstrated in a systematic study by Bajad et al. (38). Zwitterionic stationary phases (for example, sulphoalkylbetaine, Figure 1[c] and [d]) are also of interest in metabolomics studies because of a large proportion of ionizable compounds in biofluids. However, similar to amine phases, zwitterionic phases present longer analysis times as a result of strong retention of ionized compounds.

Figure 1: Chromatographic separations of a representative set of metabolites belonging to different classes - amino acids, nucleosides, organic acids, and carbohydrates - using various HILIC conditions. (a): Luna HILIC (cross-linked diol groups) column, 20 mM ammonium formate at pH 3.5; (b): Luna HILIC (cross-linked diol groups) column 20 mM ammonium acetate at pH 6.0; (c): ZIC-HILIC (sulphobetaine) column, 20 mM ammonium formate at pH 3.5; (d): ZIC-HILIC (sulphobetaine) column, 20 mM ammonium acetate at pH 6.0; (e): Luna NH2 column, 20 mM ammonium acetate at pH 9.0. Analytes: (1) aspartic acid, (2) proline, (3) threonine, (4) tyrosine, (5) guanosine, (6) inosine, (7) adenine, (8) malic acid, (9) hippuric acid, (10) nicotinic acid, (11) rhamnose, (12) trehalose, and (13) maltose. HILIC–MS experiments were performed on an UltiMate 3000 RSLC LC system (Thermo/Dionex) hyphenated to a MaXis Impact HD UHR-QqTOF (Bruker) operating in the negative ESI mode. All columns were 100 mm × 2.0 mm, 3-µm (3.5-µm for ZIC-HILIC); mobile phase: buffer (A) and acetonitrile (B); flow rate was 300 µL/min with the following gradient: 95% B for 1 min, 95–5% B for 10 min, 5% B for 3 min. Standards were dissolved in pure acetonitrile and 2 µL were injected.
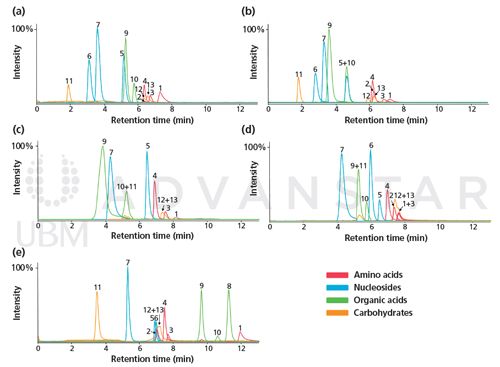
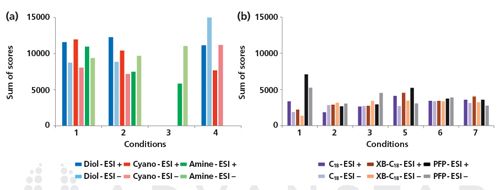
Differences between three HILIC materials, namely diol, amine, and cyano, have been compared in the context of urinary metabolic profiling by Kloos et al. for a large set of common urinary metabolites including amino acids, small carboxylic acids, carbohydrates, and nucleosides using different mobile phase compositions in both positive and negative ESI-MS mode (39). As shown in Figure 2(a), the combination of a diol column with ammonium acetate buffer at pH 7.0 and negative ESI mode led to the highest score in terms of sensitivity, peak sharpness, peak symmetry, and compound retention. Whatever the mobile phase conditions, HILIC also provided significantly better overall results than reversed-phase LC (core–shell C18, C18 with di-isobutyl side chains, and pentafluorophenyl [PFP]) for the investigated compounds (Figure 2[b]); these findings are in-line with other comparative studies (40,41). Therefore, a diol phase combined with ammonium acetate buffer at a pH of approximately 6–7 appears to give the optimal separation conditions in the context of untargeted metabolomics. In the case of unsatisfactory results, experimental parameters (stationary phase, as well as mobile phase composition, ionic strength, and pH) have to be tested to select the adequate conditions adapted to the purpose of the study. In addition to the documentation provided by manufacturers, useful guidelines and comprehensive studies are available to help in method development and selection of optimal experimental conditions for non-experienced users (38,39,42–45).

Figure 2: Sum of scores obtained under different experimental conditions in positive and negative ESI modes. (a): HILIC experiments with diol (blue), cyano (red), and amine (green) stationary phases; (b): reversed-phase LC experiments with core–shell C18 (purple), core–shell XB-C18 (C18 with di-isobutyl side chains, brown), and core–shell pentafluorophenyl (PFP, grey) stationary phases. The scores represent the chromatographic performance and were calculated based on the sensitivity (signal-to-noise ratio), peak sharpness, peak symmetry, and retention time. See (39) for further details. Experimental conditions: (1) acetonitrile/0.1% formic acid, (2) acetonitrile/20 mM ammonium acetate at pH 4, (3) acetonitrile/20 mM ammonium acetate pH 9, (4) acetonitrile/20 mM ammonium acetate pH 7, (5) methanol/0.1% formic acid, (6) methanol/20 mM ammonium acetate at pH 4, and (7) methanol/20 mM ammonium acetate pH 9. Adapted and reproduced with permission from Journal of Chromatography B 927, D.P. Kloos, H. Lingeman, W.M. Niessen, A.M. Deelder, M. Giera, and O.A. Mayboroda, Evaluation of different column chemistries for fast urinary metabolic profiling, 90–96 (2013) © Elsevier.
As a result of its complexity, HILIC is well known for a higher retention time variability compared to reversed-phase LC, mainly explained by inter-day drifts in pH or ionic strength of the buffer used (46,47). On the other hand, the effect of the column temperature seems to have a rather limited effect on the retention of hydrophilic compounds (18,19,46). During the preparation of the aqueous buffer it is therefore crucial to ensure repeatable procedures. Both buffer concentration and pH should be carefully selected according to the compounds of interest and the stationary phase, ensuring a sufficient buffer capacity and, thus, repeatable results. Modifying the buffer concentration (commonly ≤50 mM as a result of limited salt solubility in acetonitrile) has a significant impact on electrostatic interactions, where an increase in ionic strength usually leads to disruption of ionic interactions and, thus, lower retention of ionized compounds (18). Moreover, the salt concentration modifies the thickness of the water layer, which also influences the hydrophilic partitioning and retention of uncharged compounds (18,19). As a result of their MS compatibility, the vast majority of HILIC separations in metabolomics have been performed using formate and acetate-based buffers. Ammonium-based salt buffers give better results than the corresponding acid solutions. Indeed, peak shapes for ionogenic solutes, especially basic compounds, are considerably worse when using formic acid in the buffer instead of ammonium formate (48). This peak distortion has been observed for bare silica, amide, zwitterionic, and silica hybrid phases. The presence of salt buffers improves the peak shape via increased ionic strength of the mobile phase and positively impacts the formation of the water layer on the column surface (48,49).
An adequate and repeatable buffer pH is crucial in HILIC since the separation mechanisms partly rely on the pKa of analyte and stationary phase. However, the effect of the buffer pH is difficult to predict since pH and pKa are strongly affected by the presence of organic solvents. Highly organic mobile phases are therefore expected to present different apparent pH values, which may vary significantly during the gradient (49–51). Different mobile phase pH should therefore be practically tested, especially with ionizable stationary phases.
Acetonitrile remains the optimal organic solvent for HILIC analysis because it is water-miscible and aprotic. Protic solvents such as methanol, iPA, and EtOH, as well as cyclic ethers such as tetrahydrofuran are not recommended because of competition with water for the solvation of the stationary phase, which may lead to lower retention (19). During times of acetonitrile shortage, acetone can be used instead, but its use is rather limited in combination with UV (UV absorbance of the ketone function) or MS detection (formation of condensation products) (52). In HILIC, a typical gradient should start with at least 3–5% of water to ensure a repeatable formation of the water-rich layer and should not go beyond 35–40% to avoid water saturation and, thus, disruption of the aqueous layer. However, in many metabolomics applications, this proportion of water is not sufficient to elute all the polar compounds, especially when working with ionized surfaces such as sulphoalkylbetaine and amine (Figure 1). An extended gradient with a higher proportion of buffer, up to 90–95%, is often needed to break the electrostatic interactions and allow for compound elution. In this context, the water layer is disrupted and partitioning mechanisms are reduced. The re-equilibration time should therefore be sufficiently long (corresponding to at least 10–20 column volumes) to restore the water-rich layer prior to the next injection.
The latest developments in column manufacturing have been mostly focused on sub-2-µm and sub-3-µm superficially porous particles to allow for a better separation efficiency and peak capacity, similar to reversed-phase LC (17). Most HILIC phases (bare silica, amine, amide, diol, zwitterionic, and cyano; see Table 1) also exist in UHPLC column formats, while some new phase chemistries are also available, including HILIC phases that include aromatic amines and fluorinated groups.
Columns equipped with superficially porous particles (SPPs) are also commercially available but limited to some phases (bare-silica and diol for sub-2-µm core–shell as well as bare-silica, diol, and zwitterionic for sub-3-µm core–shell particles, respectively). The kinetic performance of a sub-2-µm core–shell bare-silica column is significantly improved compared with fully porous columns, which can possibly be attributed to a superior bed homogeneity and improved thermal conductivity (53). As a result of the lower viscosity of acetonitrile, a lower back pressure (two- to threefold) is observed in HILIC compared to reversed-phase LC if a conventional gradient is applied. Combining HILIC with UHPLC technology and superficially porous particles also enables the use of higher flow rates, allowing for high-throughput analysis, which is beneficial to metabolomics studies. HILIC–UHPLC–MS has already shown its potential in some metabolomics studies (54–57) but remains rarely used with superficially porous particles (58). Extending the commercially available stationary phase diversity with sub-2-µm and sub-3-µm superficially porous particles will certainly foster the rise of HILIC in metabolomics.
Mass Spectrometric Detection
HILIC has demonstrated an increased ESI-MS sensitivity compared to reversed-phase LC as a result of the high proportion of organic modifier in the eluent. Most notably, acetonitrile shows a lower dielectric constant than aqueous buffers, leading to better droplet formation and enhanced desolvation efficiency (59). The improvement in sensitivity depends on the proportion of acetonitrile in the mobile phase, flow rate, and ESI source geometry, as well as retention of the compounds of interest. For instance, a fourfold median enhancement of sensitivity has been observed on a set of basic compounds covering a broad range of physico-chemical properties (22). Even if more than 80% of the tested compounds showed a better sensitivity in HILIC–MS, large differences were observed within the studied compound set where some analytes demonstrated an improvement factor of >100-fold; however, such improvements are also dependent on the mass analyzer used.
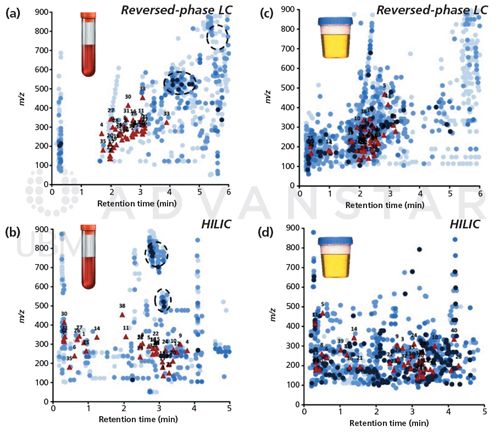
Matrix effects represent a serious challenge in metabolomics. Keeping the sample pre-treatment as simple as possible significantly increases the risk of MS signal suppression or enhancement because of co-elution of interferents with the analytes of interest. In the context of plasma analysis, HILIC has demonstrated that it is globally more prone to matrix effects than reversed-phase LC as a result of the co-elution of matrix components such as urea, creatinine, sphingomyelins, phospholipids, salts, and choline – components that are poorly (or strongly for lipids) retained in reversed-phase LC (60). Using a 2D-representation, Figure 3 illustrates the differences observed in chromatograms obtained in reversed-phase LC (BEH C18) and HILIC (bare silica) with an injection of precipitated plasma (Figure 3[a] and [b]) and diluted urine (Figure 3[c] and [d]) (61). In diluted urine, matrix effects are expected to be less prone using HILIC because of the elution of matrix components over the whole gradient compared to reversed-phase LC where interferents are clustered in certain regions of the chromatogram. Therefore, it remains difficult to draw a clear relationship between the chromatographic conditions and the prevalence of matrix effects (12,61). Hence, it is very important in metabolomics studies that matrix effects are qualitatively investigated and quantitatively assessed during method development on a representative set of metabolites, for instance using post-column infusion experiments (62) or post-extraction addition methods (63,64). The use of stable isotopically labelled internal standards is also strongly advised to ensure accurate measurements (65).

Figure 3: 2D-representations obtained for precipitated plasma and diluted urine showing m/z values as a function of retention times of endogenous compounds (blue dots) and spiked compounds of interest (red triangles). (a) and (c): Reversed-phase LC conditions with BEH C18 stationary phase and mobile phase at pH 3; (b) and (d): HILIC conditions with bare silica stationary phase and mobile phase at pH 6. (a) and (b): precipitated plasma with acetonitrile (3:1, v/v); (c) and (d): diluted urine (9:1, v/v) with water for reversed-phase LC or a solution of H2O/acetonitrile 25:75 for HILIC. MS intensity is represented by different blue shades. Black circles highlights the phospholipid region. Compounds of interest included drugs of abuse, pharmaceutical compounds, and doping agents. Experimental conditions are described in (61). Adapted and reproduced with permission from Journal of Chromatography A, A. Periat, I. Kohler, A. Thomas, R. Nicoli, J. Boccard, J.L. Veuthey, J. Schappler, and D. Guillarme, Systematic evaluation of matrix effects in hydrophilic interaction chromatography versus reversed phase liquid chromatography coupled to mass spectrometry, doi: 10.1016/j.chroma.2015.09.035 (2015) © Elsevier.
Data Analysis
As a result of the lower retention time repeatability and broader peaks obtained compared to reversed phase-LC, the data pre-processing step (including peak picking, alignment, scaling, and normalization) is of high importance in HILIC analysis to obtain a reliable matrix containing the metabolic features detected. Adequate peak picking relies on aligned chromatograms; indeed, a high variability in retention times increases the risk of wrongly assigning two chromatographic peaks to two different features in the peak-matching step. Therefore, alignment of the data is crucial and can be performed by open-source software or algorithms. For instance, algorithms developed for CE–MS data (which presents relatively high migration time variability) such as msalign2 (66) also provide appropriate results for HILIC data.
Subsequently, peak picking can be performed with open access (for example, centWave algorithms available in the XCMS package from the Scripps Institute) or commercial algorithms commonly used in reversed-phase LC–MS, bearing in mind both the larger peak widths and the multiple adducts expected (for example, acetate adducts for lipids) that are inherent to the mobile phase composition. To correct for the possible overestimation of the total number of metabolites because of multiple adduct formation, adduct-finding scripts can be used. Finally, the remaining pre-processing steps such as scaling and normalization as well as multivariate data analysis do not significantly differ from reversed-phase LC-based metabolomics approaches (67–69).
Compound identification is a critical step to convert the observed differences between populations into biological information and is known to be very challenging in reversed-phase LC-based metabolomics. A metabolite is considered identified if a minimum of two independent and orthogonal data (such as retention time and mass spectrum) has been acquired and directly compared to an authentic reference standard analyzed under identical experimental conditions (68,70). HILIC data therefore shows an additional level of difficulty because of the multiple adducts observed, which complicates the search in common databases (such as HMDB, METLIN, and KEGG). Moreover, the relatively lower column batch-to-batch repeatability represents an additional challenge because injection of authentic reference standards generally occurs at a later stage of the process, generally on a different column than the one used for sample analysis.
Applications in Untargeted Clinical Metabolomics
Few studies using HILIC in untargeted clinical metabolomics have been published so far, often in combination with reversed-phase LC to obtain the broadest possible metabolic coverage (43,71–73). Idborg et al. proposed the first metabolic fingerprinting using HILIC–MS 10 years ago on rat urine (74). In this study, urine samples were first treated by solid-phase extraction (SPE) (hydrophilic–lipophilic-based sorbent) where the wash fraction was analyzed using HILIC–MS (ZIC-HILIC stationary phase) and the eluate by reversed-phase
LC–MS, respectively. By comparing the metabolic fingerprints measured in both chromatographic modes, the authors showed the complementary nature of HILIC for the confirmation of reversed-phase LC results and its ability to detect unique variables (75). These early results were confirmed by Gika et al. who combined HILIC and UHPLC technology for the first time using a bare silica column packed with sub-2-µm particles for the analysis of obese and lean Zucker rats (76). More features (approximately 30%) were tracked with reversed-phase
LC–UHPLC than HILIC–UPHLC; this was likely a result of the sub-optimal HILIC experimental conditions. Nevertheless, based on principal component analysis, the latter allowed for the detection of important polar metabolites that were not highlighted in reversed-phase LC, demonstrating the importance of both techniques for an improved metabolic coverage.
Subsequent comparative studies clearly demonstrated the complementary nature of reversed-phase LC and HILIC in multiple and diverse applications including toxicological studies (77), renal carcinoma (78), lung cancer (79), oral squamous cell carcinoma (80), or heart failure (81), emphasizing that both techniques would bring orthogonal information and should therefore be considered together in untargeted metabolomics. Siuzdak and co-workers compared both techniques for the metabolic profiling of different samples including E. coli cells, cancer cells, and human plasma (82). For instance, they showed that an aminopropyl-based HILIC–MS method with negative ESI mode underlined key dysregulated metabolites in central carbon pathways, while reversed-phase LC–MS in positive ESI mode detected unique features belonging to lipid metabolism pathways. The two chromatographic modes were integrated into a single extraction-dual separation workflow where samples were first mixed with an 80% organic solvent mixture (methanol:acetonitrile:water 2:2:1, v/v/v) for PP prior to reconstitution in acetonitrile:water 1:1 (v/v) that allowed for dissolution of both polar and nonpolar metabolites, as well as injection on both platforms.
The future of untargeted metabolomics will likely bring more comprehensive studies systematically combining reversed-phase LC and HILIC. In this context, the on-line combination of both techniques represents an excellent possibility to offer a complete automation of the acquisition process and allow for high-throughput analysis, which is highly beneficial in large cohort metabolomics studies (83,84).
Conclusions
HILIC offers numerous advantages for the analysis of relatively polar compounds including better retention, orthogonal selectivity compared to reversed-phase LC, and improved sensitivity. Nevertheless, HILIC remains underestimated and little used in metabolomics as a result of lower retention time repeatability and poorer peak shape. These limitations, inherent to the multimodal mechanisms involved in HILIC retention, have increased the reluctance of non-experienced users for this technique. Even if HILIC is not as straightforward as reversed-phase LC, high-quality metabolomics data are ensured if good practice is followed during the entire analytical process, including (i) a careful selection of the sample diluent and volume of injection; (ii) use of ammonium-based salt buffers at a fixed and reproducible pH value and ionic strength; (iii) adaptation of the gradient profile to the stationary phase and compounds of interest; (iv) evaluation of matrix effects; and (v) careful selection of the data pre-processing parameters.
The selection of the best stationary phase in metabolomics studies also appears challenging since there is currently no equivalent to the versatile C18 used in the majority of reversed-phase LC applications. Diol phases combined with a mobile phase containing a volatile buffer at a relatively neutral pH value offer the highest performance in terms of selectivity, sensitivity, and peak shape and can thus be considered as a good starting point in metabolomics studies prior to fine tuning of the experimental conditions. The development of novel and reproducible commercially available HILIC columns, equipped with sub-2-µm fully porous or sub-3-µm superficially porous particles, will certainly encourage a more widespread use of HILIC in metabolomics as a complementary technique to reversed-phase LC to obtain the highest possible metabolic coverage.
References
- O. Fiehn, Plant Mol. Biol.48, 155–171 (2002).
- J. van der Greef, T. Hankemeier, and R.N. McBurney, Pharmacogenomics7, 1087–1094 (2006).
- J. Sun, R.D. Beger, and L.K. Schnackenberg, Personalized Medicine10, 149–161 (2013).
- R. Ramautar, R. Berger, J. van der Greef, and T. Hankemeier, Curr. Opin. Chem. Biol.17, 841–846 (2013).
- L.D. Roberts, A. Koulman, and J.L. Griffin, Lancet Diabetes Endocrinol.2, 65–75 (2014).
- T. Pacchiarotta, A.M. Deelder, and O.A. Mayboroda, Bioanalysis4, 919–925 (2012).
- M.M. Heemskerk, M. Giera, F.E. Bouazzaoui, M.A. Lips, H. Pijl, K.W. van Dijk, and V. van Harmelen, Nutrients7, 7676–7690 (2015).
- D.K. Trivedi and R.K. Iles, Biomed. Chromatogr.28, 1491–1501 (2014).
- P. Krumpochova, B. Bruyneel, D. Molenaar, A. Koukou, M. Wuhrer, W.M. Niessen, and M. Giera, J. Pharm. Biomed. Anal.114, 398–407 (2015).
- D. Kloos, H. Lingeman, O.A. Mayboroda, A.M. Deelder, W.M. Niessen, and M. Giera, TrAC61, 17–28 (2014).
- D. Rojo, C. Barbas, and F.J. Ruperez, Bioanalysis4, 1235–1243 (2012).
- L. Havlikova, H. Vlckova, P. Solich, and L. Novakova, Bioanalysis5, 2345–2357 (2013).
- P. Hemstrom and K. Irgum, J. Sep. Sci.29, 1784–1821 (2006).
- D. Kloos, R.J. Derks, M. Wijtmans, H. Lingeman, O.A. Mayboroda, A.M. Deelder, W.M. Niessen, and M. Giera, J. Chromatogr. A1232, 19–26 (2012).
- A.J. Alpert, J. Chromatogr.499, 177–196 (1990).
- W. Jian, Y. Xu, R.W. Edom, and N. Weng, Bioanalysis3, 899–912 (2011).
- L. Novakova, L. Havlikova, and H. Vlckova, TrAC63, 55–64 (2014).
- B. Buszewski and S. Noga, Anal. Bioanal. Chem.402, 231–247 (2012).
- G. Greco and T. Letzel, J. Chromatogr. Sci.51, 684–693 (2013).
- D.V. McCalley, J. Chromatogr. A 1171, 46–55 (2007).
- D.V. McCalley, J. Chromatogr. A1193, 85–91 (2008).
- A. Periat, J. Boccard, J.L. Veuthey, S. Rudaz, and D. Guillarme, J. Chromatogr. A1312, 49–57 (2013).
- A. Periat, I. Kohler, A. Bugey, S. Bieri, F. Versace, C. Staub, and D. Guillarme, J. Chromatogr. A1356, 211–220 (2014).
- A. Periat, I. Kohler, J.L. Veuthey, and D. Guillarme, Advances in Pharmaceutical Analysis, Supplement from LCGC Europe26(5), 17–22 (2013).
- F.S. Mirnaghi and A.A. Caudy, Bioanalysis6, 3393–3416 (2014).
- J.R. Denery, A.A. Nunes, and T.J. Dickerson, Anal. Chem.83,1040–1047 (2011).
- H. Gika and G. Theodoridis, Bioanalysis3, 1647–1661 (2011).
- R.J. Raterink, P.W. Lindenburg, R.J. Vreeken, R. Ramautar, and T. Hankemeier, TrAC61, 157–167 (2014).
- I. Kohler and D. Guillarme, Bioanalysis6, 1255–1273 (2014).
- B. Chauve, D. Guillarme, P. Cleon, and J.L. Veuthey, J. Sep. Sci.33, 752–764 (2010).
- J. Ruta, S. Rudaz, D.V. McCalley, J.L. Veuthey, and D. Guillarme, J. Chromatogr. A1217, 8230–8240 (2010).
- D.V. McCalley, J. Chromatogr. A1217, 3408–3417 (2010).
- D.V. McCalley, J. Chromatogr. A1217, 858–880 (2010).
- D. Bakes, Principles and Practice of Bioanalysis (2nd Edition, CRC Press, Boca Raton, Florida, USA, 2008) 69–98.
- E. Tyteca, A. Periat, S. Rudaz, G. Desmet, and D. Guillarme, J. Chromatogr. A1337, 116–127 (2014).
- P. Jandera, Anal. Chim. Acta.692, 1–25 (2011).
- D. Garcia-Gomez, E. Rodriguez-Gonzalo, and R. Carabias-Martinez, TrAC47, 111–128 (2013).
- S.U. Bajad, W. Lu, E.H. Kimball, J. Yuan, C. Peterson, and J.D. Rabinowitz, J. Chromatogr. A 1125, 76–88 (2006).
- D.P. Kloos, H. Lingeman, W.M. Niessen, A.M. Deelder, M. Giera, and O.A. Mayboroda, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci.927, 90–96 (2013).
- S. Boudah, M.F. Olivier, S. Aros-Calt, L. Oliveira, F. Fenaille, J.C. Tabet, and C. Junot, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci.966, 34–47 (2014).
- A. Periat, A. Grand-Guillaume Perrenoud, and D. Guillarme, J. Sep. Sci.36, 3141–3151 (2013).
- A. Periat, B. Debrus, S. Rudaz, and D. Guillarme, J. Chromatogr. A1282, 72–83 (2013).
- K. Spagou, H. Tsoukali, N. Raikos, H. Gika, I.D. Wilson, and G. Theodoridis, J. Sep. Sci.33, 716–727 (2010).
- T. Zhang, D.J. Creek, M.P. Barrett, G. Blackburn, and D.G. Watson, Anal. Chem.84, 1994–2001 (2012).
- I. Sampsonidis, M. Witting, W. Koch, C. Virgiliou, H.G. Gika, P. Schmitt-Kopplin, and G.A. Theodoridis, J. Chromatogr. A 1406, 145–155 (2015)
- A. Kumar, J.C. Heaton, and D.V. McCalley, J. Chromatogr. A 1276, 33–46 (2013).
- Y. Guo and S. Gaiki, J. Chromatogr. A1218, 5920–5938 (2011).
- J.C. Heaton, J.J. Russell, T. Underwood, R. Boughtflower, and D.V. McCalley, J. Chromatogr. A1347, 39–48 (2014).
- D.V. McCalley, J. Chromatogr. A1411, 41–49 (2015).
- J. Barbosa, S. Hernandez-Cassou, V. Sanz-Nebot, and I. Toro, J. Pept. Res.50, 14–24 (1997).
- S. Espinosa, E. Bosch, and M. Roses, Anal. Chem.72, 5193–5200 (2000).
- J. Heaton, M.D. Jones, C. Legido-Quigley, R.S. Plumb, and N.W. Smith, Rapid Commun. Mass. Spectrom.25, 3666–3674 (2011).
- J.C. Heaton and D.V. McCalley, J. Chromatogr. A1371, 106–116 (2014).
- G. Paglia, M. Magnusdottir, S. Thorlacius, O.E. Sigurjonsson, S. Guethmundsson, B.O. Palsson, and I. Thiele, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci.898, 111–120 (2012).
- C. Virgiliou, I. Sampsonidis, H.G. Gika, N. Raikos, and G.A. Theodoridis, Electrophoresis36, 2215–2225 (2015).
- C. Ibanez, C. Simo, D.K. Barupal, O. Fiehn, M. Kivipelto, A. Cedazo-Minguez, and A. Cifuentes, J. Chromatogr. A1302, 65–71 (2013).
- J. Chen, L. Zhou, X. Zhang, X. Lu, R. Cao, C. Xu, and G. Xu, Electrophoresis33, 3361–3369 (2012).
- F. Fei, D.M. Bowdish, and B.E. McCarry, Anal. Bioanal. Chem.406, 3723–3733 (2014).
- H.P. Nguyen and K.A. Schug, J. Sep. Sci.31, 1465–1480 (2008).
- A. Ekdahl, M.C. Johansson, and M. Ahnoff, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci.923–924, 83–91 (2013).
- A. Periat, I. Kohler, A. Thomas, R. Nicoli, J. Boccard, J.L. Veuthey, J. Schappler, and D. Guillarme, J. Chromatogr. A in press, Corrected proof, doi: 10.1016/j.chroma.2015.09.035 (2015).
- R. Bonfiglio, R.C. King, T.V. Olah, and K. Merkle, Rapid Commun. Mass Spectrom.13, 1175–1185 (1999).
- B.K. Matuszewski, M.L. Constanzer, and C.M. Chavez-Eng, Anal. Chem.75, 3019–3030 (2003).
- S. Naz, M. Vallejo, A. Garcia, and C. Barbas, J. Chromatogr. A1353, 99–105 (2014).
- M.R. Mashego, L. Wu, J.C. Van Dam, C. Ras, J.L. Vinke, W.A. Van Winden, W.M. Van Gulik, and J.J. Heijnen, Biotechnol. Bioeng.85, 620–628 (2004).
- E. Nevedomskaya, R. Derks, A.M. Deelder, O.A. Mayboroda, and M. Palmblad, Anal. Bioanal. Chem.395, 2527–2533 (2009).
- M. Berg, M. Vanaerschot, A. Jankevics, B. Cuypers, R. Breitling, and J.C. Dujardin, Comput. Struct. Biotechnol. J.4, e201301002 (2013).
- W.B. Dunn, D. Broadhurst, P. Begley, E. Zelena, S. Francis-McIntyre, N. Anderson, M. Brown, J.D. Knowles, A. Halsall, J.N. Haselden, A.W. Nicholls, I.D. Wilson, D.B. Kell, and R. Goodacre, Human Serum Metabolome, Nat Protoc6, 1060–1083 (2011).
- M. Katajamaa and M. Oresic, J. Chromatogr. A1158, 318–328 (2007).
- L.W. Sumner, A. Amberg, D. Barrett, M.H. Beale, R. Beger, C.A. Daykin, T.W. Fan, O. Fiehn, R. Goodacre, J.L. Griffin, T. Hankemeier, N. Hardy, J. Harnly, R. Higashi, J. Kopka, A.N. Lane, J.C. Lindon, P. Marriott, A.W. Nicholls, M.D. Reily, J.J. Thaden, and M.R. Viant, Metabolomics3, 211–221 (2007).
- D.Q. Tang, L. Zou, X.X. Yin, and C.N. Ong, Mass Spectrom. Rev. doi: 10.1002/mas.21445 (2014).
- S. Cubbon, C. Antonio, J. Wilson, and J. Thomas-Oates, Mass Spectrom. Rev.29, 671–684 (2010).
- H.G. Gika, G.A. Theodoridis, R.S. Plumb, and I.D. Wilson, J. Pharm. Biomed. Anal.87, 12–25 (2014).
- H. Idborg, L. Zamani, P.O. Edlund, I. Schuppe-Koistinen, and S.P. Jacobsson, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci.828, 9–13 (2005).
- H. Idborg, L. Zamani, P.O. Edlund, I. Schuppe-Koistinen, and S.P. Jacobsson, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci.828, 14–20 (2005).
- H.G. Gika, G.A. Theodoridis, and I.D. Wilson, J. Sep. Sci.31,1598–1608 (2008).
- K. Spagou, I.D. Wilson, P. Masson, G. Theodoridis, N. Raikos, M. Coen, E. Holmes, J.C. Lindon, R.S. Plumb, J.K. Nicholson, and E.J. Want, Anal. Chem.83, 382–390 (2011).
- L. Lin, Z. Huang, Y. Gao, X. Yan, J. Xing, and W. Hang, J. Proteome Res.10, 1396–1405 (2010).
- Q. Yang, X. Shi, Y. Wang, W. Wang, H. He, X. Lu, and G. Xu, J. Sep. Sci.33, 1495–1503 (2010).
- Q. Wang, P. Gao, X. Wang, and Y. Duan, Sci. Rep.4, 6802 (2014).
- M. Sun, Y. Miao, P. Wang, L. Miao, L. Liu, and J. Liu, Chromatographia77, 249–255 (2014).
- J. Ivanisevic, Z.J. Zhu, L. Plate, R. Tautenhahn, S. Chen, P.J. O’Brien, C.H. Johnson, M.A. Marletta, G.J. Patti, and G. Siuzdak, Anal. Chem.85, 6876–6884 (2013).
- D. Garcia-Gomez, E. Rodriguez-Gonzalo, and R. Carabias-Martinez, Biomed. Chromatogr.29, 1190–1196 (2015).
- Y.S. Ling, H.J. Liang, M.H. Lin, C.H. Tang, K.Y. Wu, M.L. Kuo, and C.Y. Lin, Biomed. Chromatogr.28, 1284–1293 (2014).
Isabelle Kohler received her PhD in pharmaceutical sciences in 2013 at the School of Pharmaceutical Sciences, University of Geneva in Switzerland, based on the implementation of CE–MS in clinical and forensic toxicology. She is currently carrying out her postdoctoral fellowship at the Leiden University Medical Center (LUMC) in the Center for Proteomics and Metabolomics under the supervision of Oleg Mayboroda and Martin Giera. Her main focus is the implementation and use of state-of-the-art technologies, especially HILIC–MS, in various projects linked to clinical metabolomics, notably in migraine research.
Rico Derks studied at the Vrije Universiteit in Amsterdam, The Netherlands, and received his MSc in analytical chemistry. He obtained his PhD in 2006 at the Vrije Universiteit working on the coupling of biochemical assays to mass spectrometry for detection and identification of bioactive compounds. After a postdoctoral position at the Department of Parasitology (LUMC), he is currently a Senior Researcher at the Center for Proteomics and Metabolomics (LUMC) working in the field of untargeted and targeted metabolomics.
Martin Giera obtained his PhD in 2007 in pharmaceutical chemistry at the Ludwig-Maximilians-Universität in Munich, Germany. After a postdoctoral stay and an assistant professor position at the Vrije Universiteit Amsterdam he was a visiting scientist at Harvard Medical school and became Assistant Professor at the LUMC in 2012, being responsible for the group lipid analysis and biology. Since 2015, he has been the head of the metabolomics group at the Center for Proteomics and Metabolomics (LUMC). His main research interests lie in the development of novel analytical techniques and applications for clinical and fundamental disease-related research.



















