UHPLC Tips and Tricks
An essential guide to getting the most from your UHPLC system.
Now that several years have passed since the introduction of ultrahigh-pressure liquid chromatography (UHPLC), I think we all can agree that it is a technique that is here to stay. I'm not convinced that UHPLC will be replacing all LC methods in the near future. However, UHPLC has moved beyond the research and development environment into more routine use in many companies. Some companies use UHPLC for the development of analytical methods and then covert them to conventional LC methods for routine use; others use UHPLC in all areas for new applications; others are still staying with the traditional LC methods and equipment.
Through contacts in my teaching and consulting activities, attendance at scientific meetings and e-mail with readers like you, I have collected a number of tips and techniques that are used in practical applications to get the most out of UHPLC equipment. It is this subject that I would like to explore a bit in this month's "LC Troubleshooting". I will add more to this in the future, so if you have found particular tricks that you would like to share with other readers, drop me an e-mail (John.Dolan@LCResources.com) and include "UHPLC" in the subject line. For the present discussion, I will make a binary division. LC (or as many call it, high performance LC or HPLC) will refer to equipment that operates up to the conventional pressure limits of 6000 psi (400 bar); UHPLC will refer to all equipment that operates at higher pressures.
Cleanliness
As is summarized in Table 1, the passageways through the typical UHPLC system are smaller than those for conventional LC systems. To minimize dispersion problems when 5 µm diameter particles are used as a column packing, connecting tubing should be no larger than 0.175 mm i.d. in the areas where the sample travels: between the autosampler and column and between the column and detector. If 3 µm particles are used, 0.125 mm tubing is required. Even this latter tubing can be a challenge. I remember when Hewlett-Packard first introduced their 1190 system, which was one of the early low-dispersion LC systems. Originally it was plumbed with 0.125 mm tubing, but after numerous problems with tubing becoming blocked in routine operation, they had their service engineers replace the 0.125 mm tubing with 0.175 mm i.d. tubing, which is less prone to blockage. The further challenges of UHPLC with sub 2 µm particles require even less extracolumn peak broadening, which means that even smaller tubing diameters are used; typically 0.00625 mm i.d., which is even more prone to blockage.



Table 1: LC versus UHPLC particulate challenges
One of the complaints I commonly hear about 3 µm particle diameter columns is that they become blocked much more easily than 5 µm columns. The primary reason for this is that 5 µm columns use 2 µm porosity frits to hold the particles in the column. This frit size is not sufficiently small to retain all the particles in many 3 µm particle columns, so they typically use 0.5 µm porosity frits on the outlet and either 0.5 µm or 2 µm frits on the column inlet. With sub-2 µm particles, the 0.5 µm frit is not satisfactory, so 0.2 µm frits are commonly used. Those of you who have had a microbiology class along the way will remember that 0.22 µm porosity filters are used to remove bacteria from solution. Ever see an analytical chemistry laboratory that is bacteria-free?
So how do these items affect the practical use of UHPLC in the laboratory? With 3 µm or 5 µm particles, sample filtration often can be avoided by simply placing the samples in a benchtop centrifuge, turning the speed up to the maximum for 5 min to settle any particles, and transferring the supernatant to the injection vial. As a safety measure, I strongly recommend using an 0.5 µm porosity in-line filter between the autosampler and guard column or analytical column to catch the occasional sample particle that was missed in the centrifugation step. With sub-2 µm columns and 0.2 µm frits, the centrifugation step might not be sufficient. If this is the case, every sample must be filtered through a 0.2 µm porosity frit — a very slow process. Whether centrifugation or filtration is used, a 0.2 µm porosity in-line filter is strongly recommended as a safety precaution.
Another source of particulate matter that can block the column is the mobile phase. Conventional LC applications with 3 µm or 5 µm particle columns are fairly tolerant to minor levels of mobilephase contamination. For example, if the buffer or other aqueous mobile phase is filtered through a 0.5 µm porosity solvent filter, it can be used for at least a week in most laboratories, and many laboratories extend this to two weeks. For UHPLC applications, many workers observe sufficient microbial growth in aqueous mobile phases that fresh buffers must be made daily and filtration through a 0.2 µm porosity filter is required. In either case, the reservoir should be replaced with a clean one each time the mobile phase is replenished, so that a minor microbial contamination does not amplify itself in the next batch of mobile phase.
Still another source of particulate matter is the wear of piston seals and injection valve rotors. I recently saw a UHPLC presentation that discussed the use of a continuous seal wash to help keep the piston cool so that the heat generated by the seal gripping the piston did not cause the seal to melt. The data also showed a reduction of piston seal lifetimes of fourfold at the highest pressures tested. Similarly, the increased pressure of the UHPLC system requires that the rotor seal and stator of the injection valve be pushed together more tightly, increasing friction and reducing the seal lifetimes. So it is imperative to pay close attention to system software that counts piston and injection cycles so that seals can be changed on a timely basis to minimize particles from these sources.
What does all this tell us? As one of my friends terms it, "chromatographic hygiene" must be at a much more stringent level for UHPLC than for LC applications. But many workers take care to keep particulate matter out of the system and can use UHPLC reliably in routine analysis with great results.
Pressure Effects
When most of us think about the increased pressure that is observed with a UHPLC system, it seems logical that it results from smaller-particle columns and higher linear velocities. We get faster separations at higher pressure and don't think much else about it as long as the equipment can handle the increased pressure. However, it isn't quite so simple. It has been known for more than 40 years1,2 that pressure can change the molar volume of solutes; this can result in changes in selectivity, or relative peak spacing in the chromatogram. I know that this came as a surprise to me when we were testing one of the first UHPLC systems. We observed that the peak spacing for a closely eluted triplet changed dramatically as we changed the pressure from 5400 psi (370 bar) to 12800 psi (880 bar).
With the right compounds, changes in peak spacing can be very dramatic. In the example of Figure 1,3 a change in column pressure was achieved by adding a restriction capillary between the column and detector; the same flow rate and other conditions were used. You can see that as the pressure was increased from 43 bar (625 psi) to 739 bar (10700 psi), there are two peak reversals. Peaks 3 and 4 (diphenhydramine and acetophenone) reversed retention order, as did peaks 5 and 6 (protriptyline and nitrobenzene). These changes are attributed primarily to a differential change in the molar volume of the solutes.3


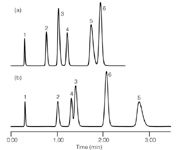
Figure 1: Pressure selectivity effects: Separation observed at (a) 43 bar and at (b) 739 bar by the addition of a restriction capillary after the column. Column: C18; mobile phase: acetonitrileâphosphate. Peaks: 1 = thiourea, 2 = propranolol, 3 = diphenhydramine, 4 = acetophenone, 5 = protriptyline, 6 = nitrobenzene. Adapted from reference 3.
What does this mean in practical terms? With the relatively small changes in pressure encountered in conventional LC systems (for example, a factor of two when the flow rate is doubled), most of us don't ever notice this effect. However, one of the common deployments of UHPLC technology in the pharmaceutical industry takes advantage of the speed of UHPLC to develop new methods in the research laboratory more quickly than could be done with conventional LC equipment. But there also is a realization that many methods are performed in a production environment where the equipment, skills and environmental conditions might not be as favourable. So to improve the chances of success, the UHPLC methods are converted to conventional LC methods on 3 µm or 5 µm particle columns that can be operated on existing, lower-pressure equipment. This can save in training, equipment costs and method downtime. However, when pressure selectivity is taken into account, scaling a method from UHPLC to LC might not be as simple as initially thought. This is of special concern for methods that must be capable of detecting many small peaks, such as stability-indicating or impurity methods. This does not mean that the UHPLC development and LC application strategy cannot work, but it does mean that special care should be taken to check for possible changes in peak spacing when the system pressure is changed. For a nice discussion of other effects of pressure in LC applications, see the introduction to reference 3.
Frictional Heating
When liquid passes through a bed of particles, the friction between the two phases creates heat (sometimes called viscous heating). As with pressure effects, with the 3 µm or 5 µm particles and 1–2 mL/min flow rates used with conventional LC methods, we seldom notice this effect. However, when the particle sizes are reduced to < 2 µm and flow rates (or more properly, linear velocities) are increased, a significant amount of heat can be generated. As has been discussed here before (for example, in reference 4), a change in column temperature can have a dramatic effect on the separation. In fact, peak reversal can occur from changes in column temperature. The use of temperature can be a powerful tool to help optimize a separation, but it also can be the source of problems in a separation if it is not carefully controlled. The effect of temperature on peak shape is shown in the illustrations of Figure 2. In Figure 2(a), a fully temperature-equilibrated column is shown. The incoming mobile phase, column bed and column oven are all at 70 °C. The uniform temperature distribution across the column means that all the molecules in a single band travel at the same speed, no matter where they are relative to the column diameter and a relatively narrow peak results.


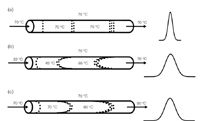
Figure 2: Effect of uneven column heating: (a) column and mobile phase fully equilibrated, (b) cold incoming mobile phase, (c) frictional heating within column.
If solvent preheating is ignored, the incoming mobile phase can be at a significantly lower temperature than the rest of the system, as shown in Figure 2(b). Here, the solvent at the centre of the column is at a lower temperature than that at the edges of the column, where the column oven has its strongest influence. As a result, molecules in the centre of the column travel more slowly than their warmer siblings closer to the column wall. This causes peak broadening. The opposite can happen if heat is created within the column, as is illustrated in Figure 2(b). Here, the centre of the column is warmer than the edges, where heat is dissipated into the column wall and oven, so the molecules in the centre travel more quickly than the ones at the edge. Once again, peak broadening results.
The combination of these temperature effects can be interesting. In one case, we noted that a sub-2 µm column generated the narrowest peaks (highest plate number) at 65 °C. Upon further investigation, we found that the oven was not performing very well, so that most of the column heating was provided not by the column oven, but by the preheated solvent (the oven was, in fact, acting as a radiator to cool the column). So under these conditions, the solvent coming into the column was warmer than that leaving (and at the column walls). On the other hand, the small particle size and high linear velocity created heat within the column, so the temperature at the column outlet was higher than at the inlet. This also caused peak broadening. But at 65 °C, the cooling effect of a poorly performing oven and the heating effect of frictional heating balanced each other and the column temperature was uniform, resulting in the best performance. Although this was an interesting observation, it certainly is not conducive to easily transferred methods. It is very important to preheat the mobile phase for methods operating above ≈40 °C, to have a column oven that works well, and to pay attention to possible influences of frictional heating.
Conclusions
UHPLC can be a great tool to speed method development and routine analysis. However, it is not without potential problems. In this discussion, we saw that cleanliness is extremely important to reliable operation. Solvents should be filtered and changed more often than with conventional LC systems. More attention may need to be given to preventive maintenance of pump and injection rotor seals. Care should be taken to minimize particulate matter in samples either through centrifugation or filtering. Additional protection of the column should be undertaken by installing a 0.2 µm in-line filter just downstream from the autosampler in every system.
We also reviewed two phenomena that can be a surprise when using UHPLC. Pressure selectivity is seldom observed with conventional LC methods, whereas it might be more prevalent in UHPLC, especially when UHPLC methods are scaled to lower pressures for routine operation. Similarly, frictional heating is not a common problem under conventional LC conditions with 3 µm or 5 µm particles, but can potentially change selectivity and column efficiency with UHPLC.
As my mother always told me when I was learning to drive as a teenager, "Be careful. Pay attention to what you are doing." This still is good advice to her chromatographer son decades later.
"LC Troubleshooting" editor John Dolan is vice president of LC Resources, Walnut Creek, California. He is also a member of LCGC Europe's editorial advisory board. Direct correspondence about this column should go to "LC Troubleshooting", Poplar House, Advanstar Communications, Park West, Sealand Road, Chester, CH1 4RN, UK, or e-mail the editor Alasdair Matheson at amatheson@advamstar.com
References
1. J.C. Giddings, Sep. Sci., 1, 73 (1966).
2. J.C. Giddings, Dynamics of Chromatography, Part I (Dekker, New York, 1965).
3. M.M. Fallas et al., J. Chromatogr. A, 1209, 195–205 (2008).
4. J.W. Dolan, LCGC North America, 25(5), 448–452 (2007).

")




")




Extracting Estrogenic Hormones Using Rotating Disk and Modified Clays
April 14th 2025University of Caldas and University of Chile researchers extracted estrogenic hormones from wastewater samples using rotating disk sorption extraction. After extraction, the concentrated analytes were measured using liquid chromatography coupled with photodiode array detection (HPLC-PDA).




Polysorbate Quantification and Degradation Analysis via LC and Charged Aerosol Detection
April 9th 2025Scientists from ThermoFisher Scientific published a review article in the Journal of Chromatography A that provided an overview of HPLC analysis using charged aerosol detection can help with polysorbate quantification.

Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.

