Sample Preparation Strategies for Characterizing RNA Modifications in Small-Volume Samples and Single Cells


This article describes challenges and opportunities in separation science in the rapidly emerging field of RNA modifications. Recent advances in sample preparation have permitted the analysis of RNA modifications in single cells by liquid chromatography–mass spectrometry (LC–MS). The sample handling and microextraction methods described here pave the way for new insights into the function of these unusual molecules in the brain and biological systems.
Apart from the four canonical nucleotides of ribonucleic acid (RNA), which are adenine (A), cytosine (C), guanine (G), and uracil (U), an extensive collection of nucleobase and ribose modifications exists that substantially diversifies the RNA code. Nearly 30 classes of modifications (~150 unique ribonucleosides) are enzymatically deposited on RNA biopolymers ranging in complexity from methylations to multistep transformations that append low molecular weight metabolites to genetically encoded RNAs (1–3). Post-transcriptional modifications influence numerous biophysical properties of cellular RNAs, including folding (4), stability (5), and translation efficiency (6,7). However, a prerequisite to understanding the functional roles of RNA modifications is the development of robust analytical methods that are capable of capturing the covariation and spatiotemporal dynamics of modified nucleosides in complex biological samples.


Kevin D. Clark of the University of Illinois at Urbana-Champaign

Liquid chromatography–tandem mass spectrometry (LC–MS/MS) techniques have played a central role in quantitatively measuring dozens of RNA modifications simultaneously (8–11), and these techniques have been leveraged to monitor changes in global modification profiles and their functional consequences (12,13). In LC–MS/MS-based strategies, total RNA is isolated from the cellular material, digested to nucleosides, and subsequently injected for instrumental analysis. During RNA isolation and pretreatment steps, sample matrix constituents, such as RNA-modifying enzymes, reactive molecules, or components that strongly bind RNA, may result in experimental artifacts that alter the observed profiles of RNAs from those of the original sample. For example, conventional RNA digestion methods may result in enzymatic or spontaneous deamination (14–16), degradation of pH-sensitive modifications (17,18), or non-natural chemical amination of RNA (19). These studies highlight the importance of selecting an appropriate sample preparation strategy and suggest careful consideration of tissue- and sample-specific matrix interferences prior to downstream analysis of RNA modifications.
Sample-to-sample differences in matrix compositions are particularly relevant because of the evidence that RNA modification statuses vary across tissue types and are unequally distributed across tissue subregions, such as within the central nervous system (CNS) (20). These findings raise intriguing questions regarding whether multiple unique RNA modifications are differentially patterned in progressively smaller samples, down to the individual cell. It is well known that single cells exhibit distinct chemical profiles that are otherwise obscured by population-averaging measurements (21,22). However, although single-cell transcriptomics workflows are now routine, analogous methods are not available for RNA modifications. Described here are recent advances in sample preparation approaches that facilitate the simultaneous characterization of multiple RNA modifications in small-volume samples, down to single cells.
Biphasic Liquid Microjunction (LMJ) Extraction: Semi-Automated, Spatially Resolved Characterization of Modified Nucleosides in Tissues
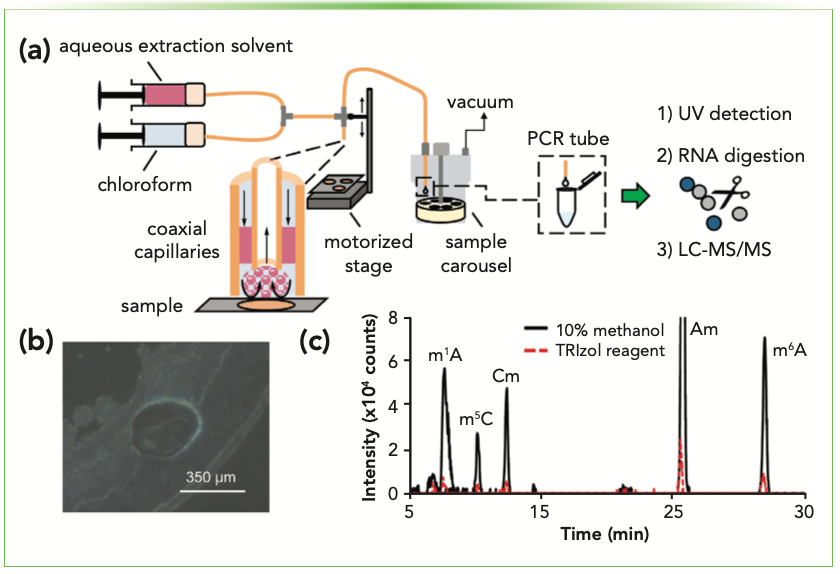
Liquid microjunction (LMJ) extraction methods leverage a microextraction probe consisting of two coaxial capillaries that facilitate the delivery and aspiration of an extraction solvent within a controlled sampling radius defined by the capillary diameter. LMJ extraction has been coupled with LC- and capillary electrophoresis (CE)–MS instrumentation, either directly or indirectly, to profile soluble sample constituents from surfaces and tissues (23,24). The optimization of solvent flow rates and precise manipulation of the extraction probe position provides a platform suitable for spatially resolved chemical analysis.
To overcome the challenge of isolating RNA from complex sam- ple matrices such as CNS tissues, the LMJ system was recently refined to include two immiscible solvents that impinge on cells or tissues with a spatial resolution of ~350 μm (Figure 1) (25). The biphasic LMJ system provided an aqueous layer where polar nucleic acids partitioned as well as an organic layer that extracted interfering compounds. Following digestion and LC–MS/MS analysis of an extract from ~500 cells, nearly a dozen RNA modifications were detected, compared to just two modifications when the same analysis was conducted with conventional liquid–liquid extraction (LLE). By integrating a rotating carousel for sample deposition, the biphasic LMJ technique represents a semi-automated approach to spatially resolved profiling of modified RNAs in complex samples.

FIGURE 1: (a) Depiction of the biphasic liquid microjunction (LMJ) RNA extraction approach. (b) Sampling footprint. (c) Representative extracted ion chromatograms for modified nucleosides obtained from biphasic LMJ extraction using different extraction solvents. Adapted with permission from reference (25).
Profiling RNA Modifications in Single Cells
The ideal sample preparation method for the analysis of RNA modifications provides a platform capable of isolating sufficient RNA material from single cells that can be digested and injected into LC–MS/MS instrumentation with minimal sample loss. One approach described by Huang and coworkers integrates sample preparation steps such as cell capture, lysis, RNA digestion, and nucleosides extraction, into a single sample tube (26). The method was successful in detecting m6A and 5-methylcytidine (m5C) in a single circulating tumor cell, representing an important step toward the characterization of multiple RNA modifications in small volumes.
Recently, an approach called single-neuron RNA modification analysis by mass spectrometry (SNRMA–MS) was developed that simultaneously detects and quantifies more than a dozen RNA modifications in individual cells (Figure 2a) (27). The method involved isolating single cells from the CNS of Aplysia californica, which were then subjected to an optimized protocol involving mechanical cell lysis, RNA denaturation, hydrolysis in an MS-compatible buffer, and LC–MS/MS. SNRMA–MS facilitated the multiplexed detection of up to 16 modified nucleosides in a single neuron, which represented approximately 60% coverage of the known RNA modification landscape in the A. californica CNS (28). In contrast, conventional phenol-chloroform RNA extraction yielded no detectable RNA modifications. These results highlighted the importance of an optimized approach for analyzing modified nucleosides in single cells.

FIGURE 2: (a) Analytical workflow for SNRMA–MS and representative extracted ion chromatograms for modified nucleosides obtained from a single neuron from the CNS of A. californica. (b) Quantitative measurement of intracellular concentrations for five RNA modifications in single neurons using SNRMA–MS. Note that *p < 0.05, **p < 0.005, paired t-test with Bonferroni−Holm correction for multiple comparisons, n = 2 animals for MCCs (four cells total), and n = 3 animals for R2/LPl1 (6 cells total). Adapted with permission from reference (27).

The detection of multiple RNA modifications by SNRMA–MS further permitted the comparison of RNA modification profiles and dynamics in identified cells from the A. californica CNS. SNRMA–MS revealed unique modification patterns for neurons with distinct physiological functions, whereas homologous cells displayed similar modifications. These differences were further investigated by implementing quantitative SNRMA–MS, which showed that 2’-O-methylation of guanosine (Gm) and cytidine (Cm) are present at significantly higher concentrations in cholinergic R2 and LPl1 motoneurons compared to serotonergic, modulatory metacerebral cells (Figure 2b). In addition to uncovering unique RNA modification profiles in single cells, SNRMA–MS has generated new questions about the functional significance of heterogeneous distribution of RNA modifications across individual cells.
Conclusions
Given the recent surge in studies correlating RNA modification dynamics in multiple tissues to biological function, it is critical to identify the appropriate sample preparation conditions that increase reliability, coverage, and detection limits for analyzing RNA modifications. Moreover, because it is increasingly evident that tissues are comprised of heterogeneous cells with unique chemical profiles, the analytical strategies used for characterization of modified RNAs must be sufficiently sensitive to detect modified nucleosides in small-volume samples. The approaches described herein are expected to advance the field of RNA modifications by contributing new knowledge of cell-specific post-transcriptional regulation. Methods capable of mapping RNA modifications to their native sequence while retaining single-cell resolution are anticipated to provide unprecedented information about the function of modified RNAs. Improvements to the efficiency and selectivity of RNA isolation from single cells via new materials for RNA separations, small-volume liquid handling approaches, and microfluidic devices will undoubtedly support these efforts and help to unravel the functional consequences of RNA modification.
References
(1) P.A. Limbach, P.F. Crain, and J.A. McCloskey, Nucleic Acids Res. 22(12), 2183–2196 (1994).
(2) W.A. Cantara, P.F. Crain, J. Rozenski, J.A. McCloskey, K.A. Harris, X. Zhang, et al, Nucleic Acids Res. 39(s1), D195–D201 (2011).
(3) P. Boccaletto, et al, Nucleic Acids Res. 46(D1), D303–D307 (2017).
(4) M. Helm, Nucleic Acids Res. 34(2), 721–733 (2006).
(5) S. Kimura and M.K. Waldor, Proc. Natl. Acad. Sci. 116(4), 1394–1403 (2019).
(6) R. Shanmugam, J. Fierer, S. Kaiser, M. Helm, T.P. Jurkowski, and A. Jeltsch, Cell Discovery 1(1), 1–10 (2015).
(7) V.A.N. Rezgui, K. Tyagi, N. Ranjan, A.L. Konevega, J. Mittelstaet, M.V. Rodnina, et al, Proc. Natl. Acad. Sci. 110(30), 12289–12294 (2013).
(8) M. Basanta-Sanchez, S. Temple, S.A. Ansari, A. D’Amico, and P.F. Agris, Nucleic Acids Res. 44(3), e26–e26 (2016).
(9) S. Kellner, A. Ochel, K. Thüring, F. Spenkuch, J. Neumann, S. Sharma, K.-D. Entian, et al, Nucleic Acids Res. 42(18), e142–e142 (2014).
(10) M. Heiss, V.F. Reichle, and S. Kellner, RNA Biol. 14(9), 1260–1268 (2017).
(11) C. Sun, M. Jora, B. Solivio, P.A. Limbach, and B. Addepalli, ACS Chem. Biol. 13(3), 567–572 (2018).
(12) C.T.Y. Chan, M. Dyavaiah, M.S. DeMott, K. Taghizadeh, P.C. Dedon, and T.J. Begley, PLoS Genet. 6(12), e1001247 (2010).
(13) C.T.Y. Chan, Y.L.J. Pang, W. Deng, I.R. Babu, M. Dyavaiah, T.J. Begley, and P.C. Dedon, Nat. Commun. 3(1), 1–9 (2012).
(14) P.F. Crain, Method. Enzymol. 193, 782–790 (1990). DOI: 10.1016/0076- 6879(90)93450-y.
(15) K. Borland, J. Diesend, T. Ito-Kureha, V. Heissmeyer, C. Hammann, A.H. Buck, et al, Genes 10(1), 26 (2019).
(16) M.S. Lowenthal, E. Quittman, and K.W. Phinney, Anal. Chem. 91(22), 14569–14576 (2019).
(17) X.-J. You, T. Liu, C.-J. Ma, C.-B. Qi, Y. Tong, X. Zhao, et al, Anal. Chem. 91(16), 10477–10483 (2019).
(18) K. Miyauchi, S. Kimura, and T. Suzuki, Nat. Chem. Biol. 9(2), 105–111 (2013).
(19) M. Jora, K. Borland, S. Abernathy, R. Zhao, M. Kelley, S. Kellner, et al, Angew. Chem. Int. Ed. 60(8), 3961–3966 (2021).
(20) M. Chang, et al, Open Biol. 7(9), 170166 (2017). DOI: 10.1098/rsob.170166
(21) J. Eberwine, H. Yeh, K. Miyashiro, Y. Cao, S. Nair, R. Finnell, et al, Proc. Natl. Acad. Sci. 89(7), 3010–3014 (1992).
(22) S.S. Rubakhin, E.V. Romanova, P. Nemes, and J.V. Sweedler, Nat. Methods 8(4), S20–S29 (2011).
(23) G.J. Van Berkel and V. Kertesz, Rapid Commun. Mass Spectrom. 27(12), 1329–1334 (2013).
(24) T.J. Comi, M.A. Makurath, M.C. Philip, S.S. Rubakhin, and J.V. Sweedler, Anal. Chem. 89(14), 7765–7772 (2017).
(25) K.D. Clark, M.C. Philip, Y. Tan, and J.V. Sweedler, Anal. Chem. 92(18), 12647–12655 (2020).
(26) W. Huang, C.-B. Qi, S.-W. Lv, M. Xie, Y.-Q. Feng, W.-H. Huang, and B.-F. Yuan, Anal. Chem. 88(2), 1378–1384 (2016).
(27) K.D. Clark, S.S. Rubakhin, and J.V. Sweedler, Anal. Chem. 93(43), 14537–14544 (2021).
(28) K.D. Clark, C. Lee, R. Gillette, and J. V. Sweedler, ACS Cent. Sci. 7(7), 1183–1190 (2021).
ABOUT THE AUTHOR
Kevin D. Clark is currently a postdoctoral fellow in Jonathan Sweedler’s group at the University of Illinois at Urbana-Champaign, Urbana, Illinois. Dr. Clark will join Tufts University as an Assistant Professor of Chemistry in Fall 2022. Direct correspondence to: clarkkd@illinois.edu and clark.kd@tufts.edu



















Advances in Non-Targeted Analysis for PFAS in Environmental Matrices
March 27th 2025David Megson from Manchester Metropolitan University in Manchester, UK, spoke to LCGC International about the latest developments in non-targeted analysis (NTA) of per- and polyfluoroalkyl substances (PFAS) in environmental matrices based on a recent systematic review paper he has collaboratively published (1).

Study Explores Thin-Film Extraction of Biogenic Amines via HPLC-MS/MS
March 27th 2025Scientists from Tabriz University and the University of Tabriz explored cellulose acetate-UiO-66-COOH as an affordable coating sorbent for thin film extraction of biogenic amines from cheese and alcohol-free beverages using HPLC-MS/MS.


Multi-Step Preparative LC–MS Workflow for Peptide Purification
March 21st 2025This article introduces a multi-step preparative purification workflow for synthetic peptides using liquid chromatography–mass spectrometry (LC–MS). The process involves optimizing separation conditions, scaling-up, fractionating, and confirming purity and recovery, using a single LC–MS system. High purity and recovery rates for synthetic peptides such as parathormone (PTH) are achieved. The method allows efficient purification and accurate confirmation of peptide synthesis and is suitable for handling complex preparative purification tasks.

