Sample Preparation Guide for Mass Spectrometry–Based Proteomics
Special Issues
A review article that summarizes the successful sample preparation strategies that have led to some of the highest peptide and protein identification rates reported in the literature
Sample preparation is an integral, but sometimes neglected, part of a successful mass spectrometry (MS)-based proteomics experiment. Good sample preparation techniques require a profound understanding of the biological samples to be analyzed and of the liquid chromatography (LC)–MS process. Depending on the experiment, biological samples often contain components (buffers and salts, detergents, polyethylene glycols, lipids, chromatins, antibodies, and streptavidin) that are not necessarily compatible with the LC–MS-MS analysis. Thus, successful sample preparation starts with a proper experimental design. Whenever possible, electrospray ionization-MS incompatible components should be systematically replaced with compatible components, such as volatile salts and MS-friendly detergents. In addition, the removal of incompatible components should be advised when they cannot be avoided (for example, an insoluble membrane protein requires detergent for solubilization or streptavidin resin is needed to enrich biotin-labeled proteins and peptides). This review article summarizes successful sample preparation strategies that led to some of the highest peptide and protein identification rates reported in the literature.
Mass spectrometry (MS)-based proteomics is playing an increasingly important role in fundamental and applied biology. As such, it attracts many newcomers to the field who want to apply this powerful technique to their specific biological question. To ensure that MS-based proteomics delivers the expected outstanding results, it is important to communicate the requirement on the sample preparation. It is also crucial to understand the unique aspects of MS measurements, next to optimizing data acquisition and interpretation. First, unlike any other biophysical technique, such as nuclear magnetic resonance (NMR) spectroscopy or microscopy and imaging, MS consumes its samples by ionizing them in the gas phase. Thus, ionizable contaminants in the sample will compete to be detected. Furthermore, complex biological matrices may not only be rich in proteins but also rich in metabolites, lipids, nucleic acids, sugars, and other molecules. If not removed, they will also compete with the peptides for ionization. In addition, complex buffer and detergents systems commonly used in biological samples can also lead to ion competition and ion suppression. Therefore, one goal should be to eliminate or at least reduce contamination and increase the number of peptides in a sample. In addition, in discovery-based proteomics operated in the data-dependent mode, where the most abundant ions are successively subjected to fragmentation, a bias toward the most abundant species exists. When one considers all these factors, it is apparent that a high demand is to be placed on sample preparation techniques when optimized MS results are sought. This review addresses some general considerations and gives practical advice for complex proteome analyses to achieve overall higher peptide and protein identifications rates (1,2).
General Guiding Principles
Although it should be noted that every proteomics sample is different and likely requires individual optimization, some general guiding principles apply to all proteomics samples. To achieve optimized results, the entire pipeline from initial sample preparation, liquid chromatography–tandem mass spectrometry (LC–MS-MS) operation, and bioinformatics analyses need to be taken into account. Systematic preventive elimination of contaminants is preferable over retrospective reduction of contaminants, although this cannot always be achieved in every setup.
To reduce the number of unwanted keratin contaminations, as a general rule, the use of a laminar air flow hood, protective clothes, and gloves is strongly encouraged for any proteomics experiments. This also means that a gel, for instance, should not be carried around uncovered when a subsequent MS investigation is intended.
Efficient cell lysis (or organelle isolation and lysis) is a prerequisite for subsequent comprehensive proteome analysis. Mechanical cell lysis is preferred over detergent-based lysis. If a detergent-based lysis is used, the detergent should be removed, either using a gel-based method or filter-assisted sample preparation (FASP) (3,4). Equally important is an effective digestion protocol that produces few missed cleavages, few unspecific cleavages, and few undesired side reactions during disulfide reduction and alkylation. Standard database searches rely on specific enzymatic cleavages, although missed cleavages and semispecific cleavages can be specified, enabling their identification as well. Nevertheless, if a peptide is present in properly cleaved and missed cleaved form, its signal intensity will be distributed into the number of forms present and the sample complexity (that is, number of detectable peptide ions) will be increased. This consideration is particularly important when very complex samples are analyzed in the smallest possible time frame.
For bottom-up proteomics approaches, trypsin is the most commonly used enzyme. Specifically, when complex proteomes are digested in solution, tryptic digestion is usually done after a predigestion with Lys-C, which is more tolerable to urea. This ensures proper digestion of proteins that would otherwise be inaccessible to trypsin. The use of urea is not without its own disadvantages and can cause carbamylations (via its decomposition to ammonium cyanate), particularly when aged solutions (that is, not prepared fresh before use) are used or when used at elevated temperatures (above 25 °C) (5–7).
While commonly used to solubilize proteins, the use of polyethylene glycol (PEG)-based detergents (NP-40, TritonX) is discouraged. As shown in Figure 1, PEG can be easily recognized in a spectrum by its equidistant spacing of 44.026 Da due to its ethoxy groups. In a worst-case scenario, this dominates a spectrum over an entire chromatogram, making the identification of peptides difficult, if not impossible. Fundamental analyses by Ogorzalek-Loo showed a limited effect of nonionic detergents such as n-dodecyl-β -D-maltoside (DDM) on MS ion suppression (8). Experience from my laboratory shows that DDM or 5-cyclohexyl-1-pentyl-β-D-maltoside (CYMAL-5) can be used instead of NP-40 or TritonX if a detergent is needed to keep proteins in solution.



Figure 1: A polyethylene glycol (PEG) detergent based spectrum is dominated by equidistant peaks, 44.026 Da apart. Each peak represents the ethoxy structure typical for PEG. Triton-X and NP-40 contain PEG structures and are commonly used in biological sample preparation. Because they ionize easily and are very hard to eliminate by biochemical or analytical means, they often have a detrimental effect on the MS outcome. It is therefore recommended to not use Triton-X or NP-40 in a proteomics sample and instead use DDM or CYMAL-5 when a detergent is deemed necessary.
Before LC–MS-MS analysis, samples need to be desalted. Depending on the sample size, commercial traps with bed volumes of as little as 0.5 µL can be used in an offline high performance liquid chromatography (HPLC) setup. When combined with UV detection, this approach is advantageous over cartridge or StageTips (9) usage, in which no quantitative data are obtained during the desalting steps.
In addition to in-solution digestion, in-gel digestion is still a common way to purify proteins from otherwise hard to remove contaminants. Direct comparison of in-gel and in-solution digestion protocols in the Mann laboratory show that the sensitivity is comparable (10). Gel digestion seems to be slightly more preferential for membrane proteins, whereas in-solution digestion seems to favor soluble proteins.
The most challenging proteomics studies are those that try to identify and quantify global proteomes of prokaryotes (such as Mycobacterium tuberculosis [11]) or eukaryotes (such as yeast [1,10,12] or HeLa lysates [13–15]). To reduce sample complexity, many experiments are focused on the subproteome of an organelle (for example, mitochondria [16]) using classical subcellular fractionation techniques. In addition, sample complexity can also be effectively reduced when enrichment techniques are used. For instance, the enrichment could be achieved using immunoprecipitations (17,18) or activity- or affinity-based pharmacoproteomic approaches (19). Enrichment could also be directed toward a specific characteristic of proteins — for example, toward newly synthesized proteins (20,21) or glycoproteins (22). For these experimental setups, additional considerations are necessary:
- In the case of organellar proteomics, all possible precautions should be taken to isolate the desired organelle. It should nevertheless be clear that all biochemical methods used to do so will remain crude methods and contaminating proteins from other cytosol or other organelles should be expected. Repeated analyses with strong statistical tests are often needed to differentiate contaminating proteins from true organellar proteins. Another (undesired) organelle can serve as a good negative control.
- In the case of enrichment proteomics, two main approaches have emerged, either immobilizing the bait or the proteins of interest. Many approaches take advantage of the strong noncovalent interactions of biotin and streptavidin using streptavidin or neutravidin beads and biotin-tagged proteins. Other commonly exploited affinity tools are the FLAG-tag, HA tag, or GFP tag used in combination with an antibody towards either of these tags. Driven by the desire to achieve the highest sensitivity, it was not too uncommon in the past to digest bound proteins directly on beads. In our experience, this has two distinct disadvantages. First, the beads themselves may introduce polymeric background ions that lead to ion suppression and may persist in the LC and MS system that could in extreme cases require the replacement of all tubing and cleaning of the ion optics. Thorough washing of the beads (before and after immobilizing the protein and without denaturing the immobilized protein) is highly recommended. Secondly, peptides generated from the digested antibody, streptavidin, or neutravidin may dominate the chromatograms and spectra, preventing lower abundance proteins to be detected. To address this disadvantage, we recommend to either elute all proteins with biotin and separate them on a gel omitting the inevitably eluting streptavidin or neutravidin band or to carefully denaturing the protein so that the bound proteins can be released predominately, followed by in-solution digestion (17). Using click-based chemistry, in which alkyne-activated agarose resins are reacted with azidohomoalanine (AHA) containing newly synthesized proteins, Eichelbaum and colleagues (23) developed a method that entirely circumvents the streptavidin contamination. The alkyne-activated agarose resins can and must be much more rigorously washed before and after binding, further reducing contamination. In our hands, so far the FLAG tag achieved best results (17,18). In all cases, these steps have to be carefully optimized in terms of bait and sample load with appropriate positive and negative controls.
LC–MS
Evidently, for the highest identification rates not only the sample needs to be prepared in an optimized fashion, but also the LC–MS-MS system needs to be operated in an optimized fashion as well (1,2). For instance, when we tested whether the LC system should be operated in a vented column setup or loading the sample directly on the column, we found the direct loading to produce higher identification rates. The vented column setup became popular because samples are loaded on a precolumn with a higher flow rate that reduces overall loading time and allows for preconcentration and on-line desalting. At the same time, some sample loss occurs during this procedure, making it a less than ideal choice for high sensitivity setups (2). In our LC setups, we optimize the solid phase and gradients to distribute the peptides as evenly as possible throughout the chromatograms (Figure 2). We preferentially use coated tips since they produce predominately doubly charged ions, decreasing the chances to identify the same peptide twice (that is, once as doubly and once as triply charged ion) (24). The mass spectrometers are operated in such a way that the highest identification rates can be achieved by optimizing the number of microscans per MS-MS scan, the signal threshold for triggering MS-MS events, the number of MS-MS events, the automatic gain control target value (ion population) for MS and MS-MS, the maximum ion injection time for MS-MS, when to choose rapid and normal scan rate, monoisotopic precursor selection criteria, and prediction of ion injection time for a selected mass resolving power (1,2).

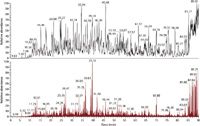
Figure 2: LCâMS (top) and LCâMS-MS (bottom) chromatogram of a global tryptic digest of a bacterium. The aim is to optimize the LC gradient so that peptides are distributed equally and are available for MS and MS-MS analysis throughout the chromatogram.
Bioinformatics
Part of our integrated approach is the use of bioinformatic quality control tools. To assess data quality and performance of an individual experiment or instrument, the freely available visualization tools LogViewer (pel.caltech.edu/software) and RawMeat (http://vastscientific.com/rawmeat/) are indispensable (24). By displaying useful instrument statistics (such as ion current fluctuation indicates problems with the spray), these tools guide users towards better sample preparation and data acquisition. Figure 3 shows the MS ion current of a 90-min analysis indicating good spray conditions with a median injection time of 3.5 ms. Only at the beginning, when no ions are eluted, higher injection times of around 100 ms are observed.

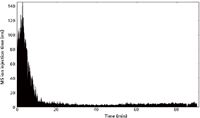
Figure 3: The MS ion injection time over retention time indicates stable spray conditions. Only at the beginning of the chromatogram where few peptides were eluted, MS ion injection times reach 100 ms and more. The median ion injection time was 3.5 ms.
Preview (http://www.proteinmetrics.com) is used to diagnose unusual modifications or incomplete digestion in a peptide mixture (25).
To convert raw files into a searchable file format, my laboratory uses ReAdw4Mascot2 because of its superior monoisotopic peak picking. To identify peptide spectrum matches, my laboratory has developed a freely available second generation ROCCIT search engine (roccit.caltech.edu), which is among the first search engines that take into account known modifications listed in Uniprot (2). The sophisticated signal processing tools for Thermo raw files make MaxQuant a premier choice for quantitative bottom-up proteomics (1,2,26).
What to Expect?
To optimize our workflow and for quality control, we use global tryptic digests of yeast lysates that we analyze in 160-in analyses. In an optimized analysis using an Orbitrap Elite, from a yeast lysate (200 ng sample load), we expect 5500 MS scans and 41,000 MS-MS scans yielding 17,000 peptide spectrum matches and identifying 12,800 unique peptides (or 2100 proteins with more than two peptides) (2). This equals 80 unique peptides and 13 proteins/min analysis and is among the highest identification rates reported in the literature (2).
Conclusion
While there is no magic bullet, knowledge in the sample requirements and careful sample preparation enables high identification rates of complex biological samples. Sample preparation is an integral and often underestimated part of the success of a proteomics experiment and is as important as optimizing the LC, MS, and bioinformatics analyses. When all parts of a proteomics experiment are optimized, outstanding results can be obtained.
Acknowledgments
The Proteome Exploration Laboratory is funded by the Gordon and Betty Moore Foundation through Grant GBMF775, the Beckman Institute and the NIH award SRR029594A. I thank my current and former lab members for their contributions.
References
(1) A. Kalli and S. Hess, Proteomics 12, 21–31 (2012).
(2) A. Kalli et al., J. Proteome Res. In press, http://pubs.acs.org/doi/pdf/10.1021/pr3011588 (2013).
(3) L.L. Manza et al., Proteomics 5, 1742–1745 (2005).
(4) J.R. Wisniewski et al., Nat. Methods 6, 359–U60 (2009).
(5) G.R. Stark, Biochemistry 4, 2363–2367 (1965).
(6) G.R. Stark, Biochemistry 4, 1030–1036 (1965).
(7) L. Kollipara and R.P. Zahedi, Proteomics 13, 941–944 (2013).
(8) R.R. Loo, N. Dales, and P.C. Andrews, Protein Sci. 3, 1975–1983 (1994).
(9) J. Rappsilber, M. Mann, and Y. Ishihama, Nat. Protoc. 2, 1896–1906 (2007).
(10) L.M. de Godoy et al., Nature 455, 1251–1254 (2008).
(11) C. Bell et al., J. Proteome Res. 11, 119–130 (2012).
(12) K.J. Webb et al., J. Proteome Res. 12, 2177–2184 (2013).
(13) A. Michalski et al., Mol. Cell. Proteomics 11, O111.013698 (2012).
(14) J.V. Olsen et al., Mol. Cell. Proteomics 8, 2759–2769 (2009).
(15) D. Winter and H. Steen, Proteomics 11, 4726–4730 (2011).
(16) N.C. Chan et al., Hum. Mol. Genet. 20, 1726–1737 (2011).
(17) N.W. Pierce et al., Cell 153, 206–215 (2013).
(18) J.E. Lee et al., Mol. Cell. Proteomics 10, M110 006460 (2011).
(19) S. Hess, Proteomics Clin. Appl. 7, 171–180 (2013).
(20) J.J. Hodas et al., Proteomics 12, 2464–2476 (2012).
(21) D.C. Dieterich et al., Proc. Natl. Acad. Sci. USA 103, 9482–9487 (2006).
(22) G.T. Smith, M.J. Sweredoski, and S. Hess, Journal of Proteomics, In press, doi: 10.1016/j.jprot.2013.05.011, (2013).
(23) K. Eichelbaum et al., Nat. Biotechnol. 30, 984–90 (2012).
(24) M.J. Sweredoski et al., J. Biomol. Tech. 22, 122–126 (2011).
(25) Y.J. Kil et al. Anal Chem. 83, 5259–5267 (2011).
(26) J. Cox and M. Mann, Nat. Biotechnol. 26, 1367–1372 (2008).
Sonja Hess is with the Proteome Exploration Laboratory from the Beckman Institute at the California Institute of Technology in Pasadena, California. Direct correspondence to: shess@caltech.edu










Thermodynamic Insights into Organic Solvent Extraction for Chemical Analysis of Medical Devices
April 16th 2025A new study, published by a researcher from Chemical Characterization Solutions in Minnesota, explored a new approach for sample preparation for the chemical characterization of medical devices.



Study Explores Thin-Film Extraction of Biogenic Amines via HPLC-MS/MS
March 27th 2025Scientists from Tabriz University and the University of Tabriz explored cellulose acetate-UiO-66-COOH as an affordable coating sorbent for thin film extraction of biogenic amines from cheese and alcohol-free beverages using HPLC-MS/MS.

Multi-Step Preparative LC–MS Workflow for Peptide Purification
March 21st 2025This article introduces a multi-step preparative purification workflow for synthetic peptides using liquid chromatography–mass spectrometry (LC–MS). The process involves optimizing separation conditions, scaling-up, fractionating, and confirming purity and recovery, using a single LC–MS system. High purity and recovery rates for synthetic peptides such as parathormone (PTH) are achieved. The method allows efficient purification and accurate confirmation of peptide synthesis and is suitable for handling complex preparative purification tasks.

