Reflecting on the Influence of the Current State of Sample Preparation on GC, Part 1


Sample preparation and sample injection are often the most challenging steps in a full gas chromatographic method. Gas chromatography (GC) is generally amenable to liquid and vapor phase samples, and in most applications, the sample must be in the vapor phase as it enters the column. This can be seen in the most common injection techniques, split and splitless, in which a liquid sample is injected using a syringe into an inlet where it is vaporized. Sample preparation for GC can be online, with direct interfacing, usually through a transfer line, between a separate sample preparation technique, such as static headspace extraction, or offline, with samples prepared separately, aliquoted into vials, usually as solutions, and injected as liquid samples.
Table I provides a summary of some sample preparation techniques commonly used with GC for solid, liquid, and vapor samples. Typically, for solid samples, the solid is dissolved or otherwise treated, followed by one of the techniques for liquid samples. Commonly, solid samples are dissolved in a solvent, but they can also be extracted using a number of instrumental techniques, including headspace extraction, accelerated solvent extraction, and microwave assisted extraction. Liquid and vapor samples can be injected “neat,” but usually require sample preparation to remove nonvolatile or reactive sample components.

.")
Table I: Sample types amenable to common sample preparation techniques for GC (S = solid, L= liquid, G = gas).

In LCGC surveys reported in 1991 (1) and 2023 (2), we see that sampling and sample preparation occupied over 50% of analyst time in a typical analysis, so the importance of sample preparation cannot be overlooked. In both surveys, analysts were also asked about the most important error sources in their methods, with the sample preparation-related responses of sample processing, operator and contamination combining for just over 50% of the responses in 1991 and about 37% in 2023. In 2023, calibration was stated as the largest single contributor to error, while in 1991, it was sample processing. This change was attributed to the rise in automation over the past 30 years. Interestingly, the major sources of error in most gas chromatographic methods are likely outside of the instrument, either in sample preparation and handling (which happens prior to the chromatographic analysis), or data analysis (which happens following chromatography).
Subtleties Fundamental to All Sample Preparation Techniques
From Table I, we see that nearly all sample preparation techniques used with GC involve a phase transition and phase equilibrium, just as GC depends on the theory of equilibrium between the mobile (vapor) and stationary (usually liquid or solution) phases. A brief review of phase equilibrium applied to GC and sample preparation will show principles common to all sample preparation techniques. The chemical equation and equilibrium constant expression for a phase transition, shown in equation 1, are among the simplest that describe a chemical process, but there are many subtleties.

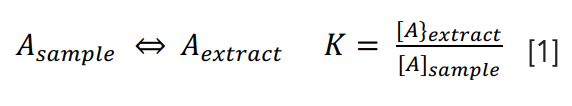
The first subtlety is the consequences of the value of the equilibrium constant. This determines how much of the analyte can be transferred from one phase to the other in a single extraction step. In GC, a single extraction step is analogous to a single theoretical plate, as we saw last year in our discussion of the classical Golay equation (3,4). Figure 1 shows a comparison of several common extraction methods, including solid-phase microextraction (SPME), liquid-liquid extraction (LLE), and low-volume LLE. The mole ratio of a hypothetical analyte extracted from an aqueous phase into an organic phase is plotted against log K. If K is large, indicating that the analyte is much more soluble in the organic phase than the aqueous phase (as is commonly seen in gas chromatographic methods, when extracting organic compounds from water or other aqueous systems into an organic phase), then a nearly complete, or quantitative, transfer of the analyte from the aqueous phase to the organic phase is possible.

Figure 1: Comparison of some extraction methods. MLLE–liquid-liquid extraction with both phases of low volume; SPME–solid-phase microextraction; High–large volume of aqueous phase extracted with a small volume of organic phase; Low–like MLLE but using a smaller volume of organic phase and a large volume of aqueous phase.
The second subtlety, also seen in Figure 1, is seen in the relative volumes of the sample (in this case aqueous) phase and the extract (organic) phase. The micro-liquid-liquid extraction (MLLE) case is a low, but roughly equal, volume of both phases, volumes that would fit into a single autosampler vial; the high case is a large volume of aqueous phase extracted with a small volume of organic phase, while the low case is like MLLE, but using a smaller volume of organic phase and a large volume of aqueous phase. Finally, SPME represents an extreme volume difference with the very low-volume coated fiber immersed in a large volume of aqueous sample.
The third subtlety is that while equilibrium constants may be very large or very small, they are finite. This means that in any extraction system, even with a very large constant, some amount of the analyte will remain in the original phase. This challenge opens an additional idea that has not been explored much in the literature. If the equilibrium constant is small, multiple extractions form the same sample, using a fresh extraction phase, can result in appreciable extraction of the analyte when the separate aliquots of the extraction phase are combined.
A fourth subtlety is that for any chemical reaction or process, especially extraction, reaching equilibrium takes time. Even in the simplest of systems, a “dilute and shoot” procedure, the diluted liquid must be mixed. When diluting one liquid by another, how does the analyst know that the two liquids have fully mixed into a homogeneous mixture? Mostly, we count on indirect evidence based on our judgment of the time and vigorousness of mixing, appearance of the solution, and reproducibility of the data. The same questions apply to simply dissolving solids. Mostly, we determine that the solid has dissolved when we no longer see particles, and we also know that no solid dissolves instantly. In short, when diluting or making solutions, allow sufficient time for complete mixing.
We see, from the simplest of techniques, that all sample preparation methods require time to come to equilibrium. This amount of time may be on the same order of or longer than the time required for the chromatographic separation. Our fifth subtlety in all sample preparation techniques is kinetics. While equilibrium determines how much analyte we can transfer between phases, kinetics determines how long it takes. Due to the variety and complexity of most samples, the kinetics of extraction must be determined experimentally and may vary widely for seemingly similar samples.
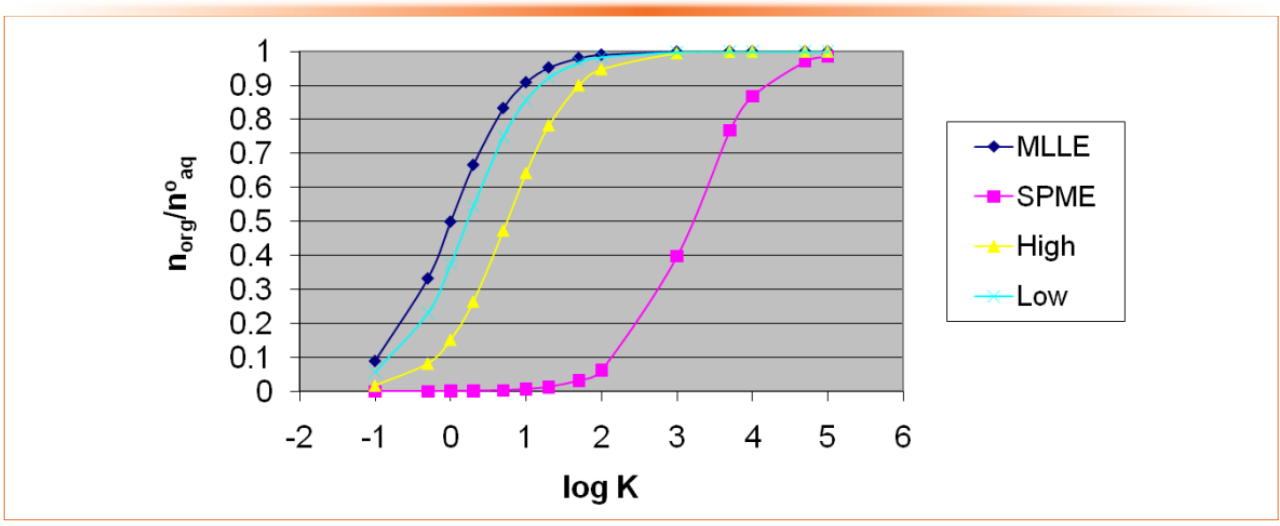
Fortunately, coming back to the simplicity of equation 1, most extraction techniques follow or approximate first order kinetics. In the simplest case, we have individual particles partitioning between two phases. Extraction kinetics may be complicated by additional matrix components in the sample phase that bind or adsorb the analytes of interest. In any method development involving extraction, the kinetics of extraction should be carefully studied. A kinetic plot for a simple and fun extraction is shown in Figure 2. This is the extraction of fats from a chocolate bar using supercritical fluid extraction (5). In this case, time is measured by the volume of supercritical carbon dioxide delivered at constant flow. This is a quantitative extraction example, but it shows the key elements of extraction kinetics.

Figure 2: Extraction of fat from a candy bar using supercritical fluid extraction. Reprinted with permission from reference (5). Copyright 1996, American Chemical Society.
For the best reproducibility and maximum recovery, extraction should be allowed to proceed for long enough so that the amount extracted is expressed on the flat, nearly horizontal part of the curve. Small variations in the extraction time or conditions will have the least effect on this portion of the curve. Shorter extraction times, where the curve is steeper, can be used with calibration, but at the risk of larger uncertainty and impact of minor variations in the extraction conditions.
A sixth subtlety is the experimental uncertainty inherent in the use of glassware for volumetric measurements and sample transfers. The most common experimental error that impacts all analytical methods, including GC, is the experimental error and overconfidence involved with any use of volumetric glassware, which is often assumed to be error-free, but is not. A second, often hidden, error is seen in the volume of the glassware itself, as pipets and flasks have increasing relative uncertainty as their volumes get smaller. This is discussed in detail in a previous column, where we see that smaller volume flasks and transfer pipets introduce larger experimental uncertainties, and that, with improved instrumentation over the decades, these uncertainties may eclipse those from the instrumentation or other method steps (6). In short, there is a tradeoff of increased experimental uncertainty when using serial dilution methods or any method that requires additional sample transfers and volume measurements, which use smaller solvent volumes and single-step dilution, which uses larger volumes.
In summary, six subtleties in sample preparation result from the seeming simplicity of equation 1.
- The equilibrium constant determines how much can be extracted.
- The relative volumes of the two phases determine how much can be extracted.
- Equilibrium constants, large or small, are finite. There is always some analyte in both phases.
- Dissolving and mixing take time.
- Extractions are subject to kinetics.
- Glassware has inherent experimental uncertainty.
Returning to the survey results, Raynie points out some interesting changes since 2003 that, along with the subtleties, provide some interesting challenges and observations about the current state and future of sample preparation for GC. In the survey, several shifts are seen since 2013, with new fields, including cannabis, cosmetics and personal care and a drop in the percentage of users reporting work in pharmaceuticals. In all industries, there are both increased regulation and pressure to “go green” with reduced solvent and consumable usage, miniaturization, more efficient laboratories along with lower detection limits and reduced experimental uncertainties (7). Interestingly, the subtleties can work against these pressures, as the kinetics of extraction often work toward longer extraction times and lower sample throughput, and the glassware uncertainty favors larger samples and more solvent usage. When evaluating techniques, users report the greatest importance in price, time, recovery and low contamination.
Sample preparation and GC are inherently connected. One cannot run a gas chromatograph without samples, and all samples require some amount of preparation, from the simplicity of loading “neat” samples into vials, through the complexity of on-line instrumental extractors, such as accelerated solvent extraction (ASE), supercritical fluid extraction (SFE), and headspace. All extraction techniques are subject to equilibrium, kinetics and experimental uncertainty, as is GC. With GC becoming simplified over the decades, the often-overlooked time, complexity, kinetics, and equilibrium involved in sample preparation may often be the most important and challenging steps in the full method.
References
(1) Majors, R. E. Trends in Sample Preparation. LCGC 1991, 9 (1), 16–20.
(2) Raynie, D. Trends in Sample Preparation, Part I: Current State of the Field. LCGC N. Am. 2023, 41 (9), 374–380, 393. DOI: 10.56530/lcgc.na.tu5672b5
(3) Snow, N. H. Is Golay’s Famous Equation for HETP Still Relevant in Capillary Gas Chromatography? Part 1: A Common View of HETP. LCGC N. Am. 2022, 40 (1), 22–26, 31. DOI: 10.56530/lcgc.na.uk4587d6
(4) Snow, N. H. Is Golay’s Famous Equation for HETP Still Relevant in Capillary Gas Chromatography? Part 2: Assumptions and Consequences. LCGC N. Am. 2022, 40 (2), 68–71. DOI: 10.56530/lcgc.na.mm7085y7
(5) Snow, N. H.; Dunn, M.; Patel, S. Determination of Crude Fat in Food Products by Supercritical Fluid Extraction and Gravimetric Analysis. J. Chem. Educ. 1996, 74 (9) 1108–1111. DOI: 10.1021/ed074p1108
(6) Snow, N. H. Is the Solution Dilution? Hidden Uncertainty in Gas Chromatography (GC) Methods. LCGC N. Am. 2022, 40 (7), 304–308. DOI: 10.56530/lcgc.na.re9187d2
(7) Snow, N. H. Green Chemistry: What Is It (and What Is It Not)? And How Does It Apply to Gas Chromatography? LCGC N. Am. 2023, 41 (5), 176–180. DOI: 10.56530/lcgc.na.az3979e4
About the Author

Nicholas H. Snow is the Founding Endowed Professor in the Department of Chemistry and Biochemistry at Seton Hall University, and an Adjuncy Professor of Medical Science. During his 30 years as a chromatographer, he has published more than 70 refereed articles and book chapters and has given more than 200 presentations and short courses. He is interested in the fundamentals and applications of separation science, especially gas chromatography, sampling, and sample preparation for chemical analysis. His research group is very active, with ongoing projects using GC, GC-MS, two-dimensional GC, and extraction methods including headspace, liquid-liquid extraction, and solid-phase microextraction. Direct correspondence to: LCGCedit@mmhgroup.com

















New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.

University of Rouen-Normandy Scientists Explore Eco-Friendly Sampling Approach for GC-HRMS
April 17th 2025Root exudates—substances secreted by living plant roots—are challenging to sample, as they are typically extracted using artificial devices and can vary widely in both quantity and composition across plant species.

Thermodynamic Insights into Organic Solvent Extraction for Chemical Analysis of Medical Devices
April 16th 2025A new study, published by a researcher from Chemical Characterization Solutions in Minnesota, explored a new approach for sample preparation for the chemical characterization of medical devices.

