Multi-Active Method (MAM) for the Analysis of Agriculture Product Technical Ingredients and Formulated Products
In the pesticide industry, regulatory laboratories are responsible for the quality of the material sold in the open market. Laboratories receive samples from the open trade that are sold in their individual countries. To this end, methodologies to analyze for the technical active ingredient (AI) and formulated products are available to them from the commercial agricultural product manufacturers’ registration dossiers. The methods are part of the regulatory package submitted to countries for approval to sell and are the official registered methods for the AI and the formulated product.
AI analytical methods are typically liquid or gas chromatography-based and identify and quantify the AI in technical material and formulated products. These methods have one purpose: to determine if the product being produced and sold meets registered specifications. The commercial producer analyzes manufactured material before it is released to the open market to ensure it meets registered specifications, and worldwide regulatory agencies use these methods to check that material that is sampled from the open market also meets registered specifications.
So just how many registered AI methods are there? There are 1,055 active pesticide ingredients authorized for use in the United States alone (1), and worldwide active ingredient numbers are proportional (2). These active ingredients represent 20,000 marketed or formulated products, many with different concentration levels of the active or mixtures of two or more actives. In a small country such as Vietnam, there are products with more than 3,000 separate trade names registered.
For manufacturers, where only one product is made at a time, the analysis of what is produced to determine the acceptability of a product is routine. Generally, many batches or a continuous process are monitored with the company method which is set up in the site laboratory and maintained throughout the manufacturing campaign. Chromatographic columns that have been chosen for this methodology are specific for the analysis at hand, and can be routinely used for years, that is, the life of the product. The cost is trivial by comparison to the expense of supporting the manufacturing process.
Such is not the case in the regulatory laboratories, where potentially hundreds of different types of sample and active ingredients can be seen every year. With each set of unique conditions provided by the manufacturers, the regulatory laboratories are faced with the decision to purchase a new and different analytical column, or not follow the enforcement company method.
Because of this, a multi-analyte method has been developed at the Irish Department of Agriculture, Food and The Marine, contributed to by the regulatory laboratories in Belgium and the Czech Republic, analyzing more than 70 active ingredients using high performance liquid chromatography (HPLC) and more than 35 active ingredients by ultrahighpressure liquid chromatography (UHPLC). The method has been designed for use by quality control laboratories and is suitable for determining a range of active substances in a wider range of formulated products as well as the technical AI itself. The method has been validated for linearity, precision, accuracy, and specificity for seven technical active ingredients as defined by FMC Corporation.
The method and results are presented in this report.
Experimental
Reagents (Reagent Grade, Except as Noted)
Analytical standards, of known purity, stored in refrigerator or as per laboratory protocol.
Dicyclohexyl Phthalate (DCHP) > 97% pure – internal standard
Ethyl Acetate, HPLC grade
Acetone, HPLC grade
Acetonitrile, HPLC grade
Water, HPLC grade
Phosphoric Acid, HPLC grade
Formic Acid, MS grade
Apparatus
A high performance liquid chromatography (HPLC) system equipped with a constant temperature column compartment, a temperature-controlled autosampler, an autosampler capable of delivering 10 μL injections, a variable-wavelength UV detector or photodiodearray detector, and a digital integrator or other data handling device.
OR
An ultrahigh-pressure liquid chromatography (UHPLC) system equipped with a constant temperature column compartment, a temperature-controlled autosampler, an autosampler capable of delivering 0.5 μL injections, a variable-wavelength UV detector or photodiode-array detector, and a digital integrator or other data handling device.
- HPLC column: Kinetex C18, 100 mm × 4.6 mm (i.d.) × 2.6 μm or equivalent.
- UHPLC column: Kinetex C18, 100 mm × 2.1 mm (i.d.) × 2.6 μm or equivalent, and precolumn Phenomenex C18, 2.1 mm i.d. or equivalent.
- Electronic integrator or data system
- Ultrasonic bath capable of being heated up to 50 °C
- Water bath
- Analytical balance
- Grinder/pulverizer
- Mechanical shaker
- Calibrated pH meter
- Organic PTFE filter, 0.22 or 0.45 μm
Standard Preparation
Internal Standard Stock Solution
Dissolve 80 mg of dicyclohexyl phthalate (to the nearest 0.1 mg) into a 100 mL volumetric flask and dilute to the mark with acetone. Internal standard solution concentration is approximately 0.08 mg/mL.
Calibration Solution
Weigh in duplicate about 10 mg of the required analytical standard to the nearest 0.1 mg in a volumetric flask (100 mL). Dissolve and fill to the mark with acetonitrile. This gives a standard concentration of approximately 100 mg/L, calibration solutions CA and CB..
Sampling
Ideally, an intact product should be taken from the market as intended for use with an end user.
Sample Preparation
Sample preparation is dependent on the formulation type being analyzed. The procedures are different for liquid and solid formulations and for technical material. Once the samples are prepared, the active substances are analyzed by either high performance or ultrahigh-pressure liquid chromatography with either UV or diode-array detection (HPLC-UV, HPLC-DAD, UHPLCUV, or UHPLC-DAD, respectively), depending on the instrumentation available.
Liquid or Suspension Formulations
Thoroughly shake the sample container to homogenize the sample before use. Weigh in duplicate (to the nearest 0.1 mg) sufficient sample to contain 9 to 11 mg of the active substance into a volumetric flask (100 mL). For suspension formulations add 5 mL of water to disperse. Subsequently, add 70 mL of acetonitrile. If it is necessary, sonicate for dissolution of active ingredient. Allow the solution to cool to ambient temperature and fill to the mark with acetonitrile (Solutions SA and SB). This gives an active ingredient concentration of approximately 100 mg/L. For UHPLC, filter samples through 0.22 μm filter.
Solid Formulations
For solid formulations, dispersion with 10% water prior to solvent addition is recommended. Weigh 15–20 g of the sample into a mortar and pestle or grinder/pulverizer and homogenize thoroughly. Subsample by weighing in duplicate, to the nearest 0.1 mg, sufficient sample to contain 9 to 11 mg of the active substance into a 100 mL volumetric flask. Add 70 mL of the internal standard solution. If it is necessary, sonicate for dissolution of active ingredient. Allow the solution to cool to ambient temperature, fill to the mark with acetonitrile, and mix well (Solutions SA and SB). This gives a sample concentration of approximately 100 mg/L. For UHPLC, filter samples through a 0.22 μm filter.
Mobile Phase Preparation
Mobile Phase A, Aqueous
Prepare by pipetting phosphoric or formic acid into a 2000 mL volumetric flask containing 1500 mL of water, add water to the 2000 mL mark, and thoroughly mix. pH should between 2.0 and 2.2.
Mobile Phase B
Acetonitrile.
HPLC Chromatographic Conditions
Column: HPLC column, Kinetex C18, 150 mm × 4.6 mm i.d. × 2.6 μm or equivalent
Flow rate: 1.0 mL/min
Injection volume: 0.5 μL
Detector wavelength: 220–340 nm. The optimum wavelength can be established by analyzing the analytical standard on PDA detector prior to analysis if enforcement method is not available for reference.
Run time: 18 min
Initial mobile phase concentration: A = 65%: 0.1% pH 2.0–2.2; o-phosphoric or formic acid-adjusted water; B = 35%: Acetonitrile
HPLC mobile phase gradient: see Table I.


Table I: HPLC Mobile Phase Gradient
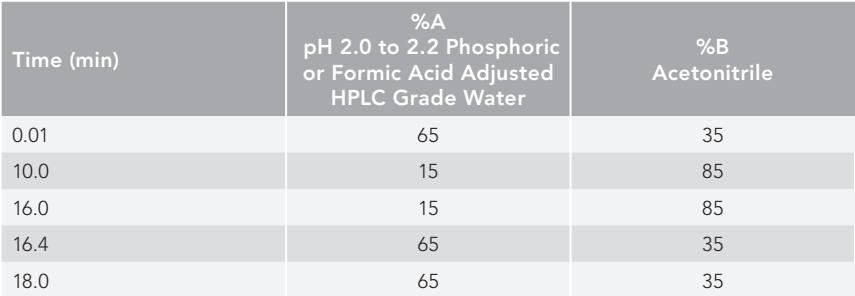
Column temperature: 25 °C
UHPLC Chromatographic Conditions
Column: Kinetex C18, 150 mm × 2.1 mm (i.d.) × 2.6 μm or equivalent, and precolumn Phenomenex C18, 2.1 mm i.d. or equivalent.
Flow rate: 0.4 mL/min
Injection volume: 0.5 μL
Detector wavelength: 210–330 nm
The optimum wavelength can be established by analyzing the analytical standard on a DAD detector prior to analysis if registration method is not available for reference.
Run Time: 8.5 min
Initial mobile phase concentration: A = 65%: 0.1% pH 2.0–2.2; o-phosphoric or formic acidadjusted water; B = 35%: Acetonitrile
UHPLC mobile phase gradient: see Table II.

Table II: UHPLC Mobile Phase Gradient
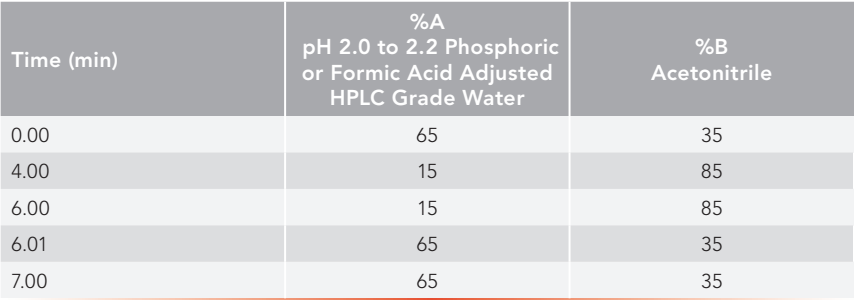
Column temperature: 25 °C
Equilibration of the System
For HPLC
Pump sufficient mobile phase through the column to equilibrate the system approximately 20 to 30 min at the recommended flow rate of 1.0 mL/min. Inject 5 µl portions of the standard solution until the response obtained from two consecutive injections deviates by less than 1.0 %, preferably less than 0.5%.
For UHPLC
Pump sufficient mobile phase through the column to equilibrate the system approximately 20 to 30 min at the recommended flow rate of 0.4 mL/min. Inject 0.5 µl portion of the standard solution until the response obtained from two consecutive injections deviates by less than 1.0% for retention times and by less than 2.0% for peak areas.
Determination of the Species of Interest
For HPLC
Inject in duplicate 5 µL portions of each sample solution, bracketing them by injections of the calibration solutions as follows; calibration solution CA, sample solution S1A, sample solution S1B, calibration solution CB, sample solution S2A, sample solution S2B, and so on. Measure the relevant peak areas.
For UHPLC
Inject in duplicate 0.5 µL portions of each sample solution, bracketing them by injections of the calibration solutions as follows; calibration solution CA, sample solution S1A, sample solution S1B, calibration solution CB, sample solution S2A, sample solution S2B, and so on. Measure the relevant peak areas.
Calculations
Calculate the mean value of each pair of response factors bracketing the two injections of a sample and use this value for calculating the active substance contents of the bracketed sample injections as shown below.
Individual response factors for CA and CB:


Mean response factor for CA and CB:


Content of active substance wt % active substance =


where:
fi = individual response factor
f = mean response factor
Hs = peak area of active substance in the calibration solution
Hw = average peak area of the active substance from the two injections of the sample solution
s = mass of the active substance working standard in the calibration solution (mg)
w = mass of sample taken (mg)
P = purity of the active substance working standard (g/kg)
Results and Discussion
The conditions that have been outlined above for both HPLC and UHPLC have been used to analyze the compounds listed in Table III and Table IV, respectively. Given in these tables are the formulation types of the samples where the method was used successfully, the UV maximum wavelength determined during our experiments, and the declared content including the units of the specific material.

Table III: Formulated products examined by HPLC conditions of the multi-analyte method
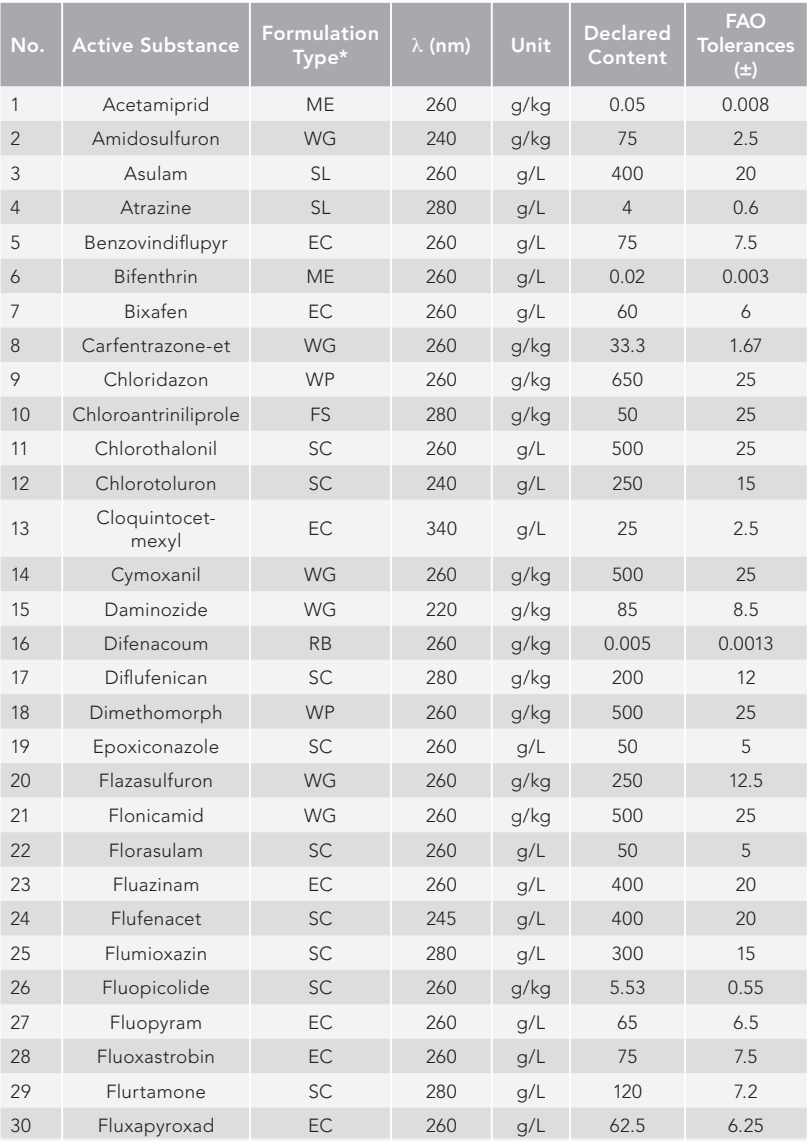

Table III, Continued: Formulated products examined by HPLC conditions of the multi-analyte method
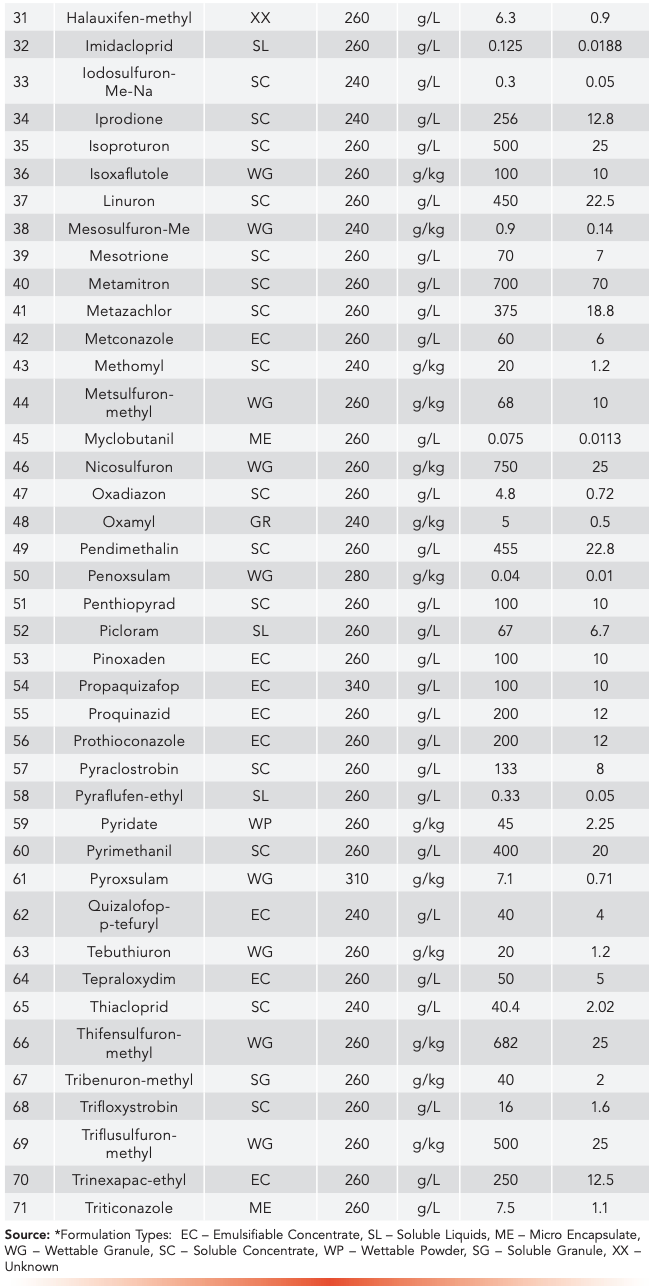

Table IV: Formulated products examined by UHPLC conditions of the multi-analyte method
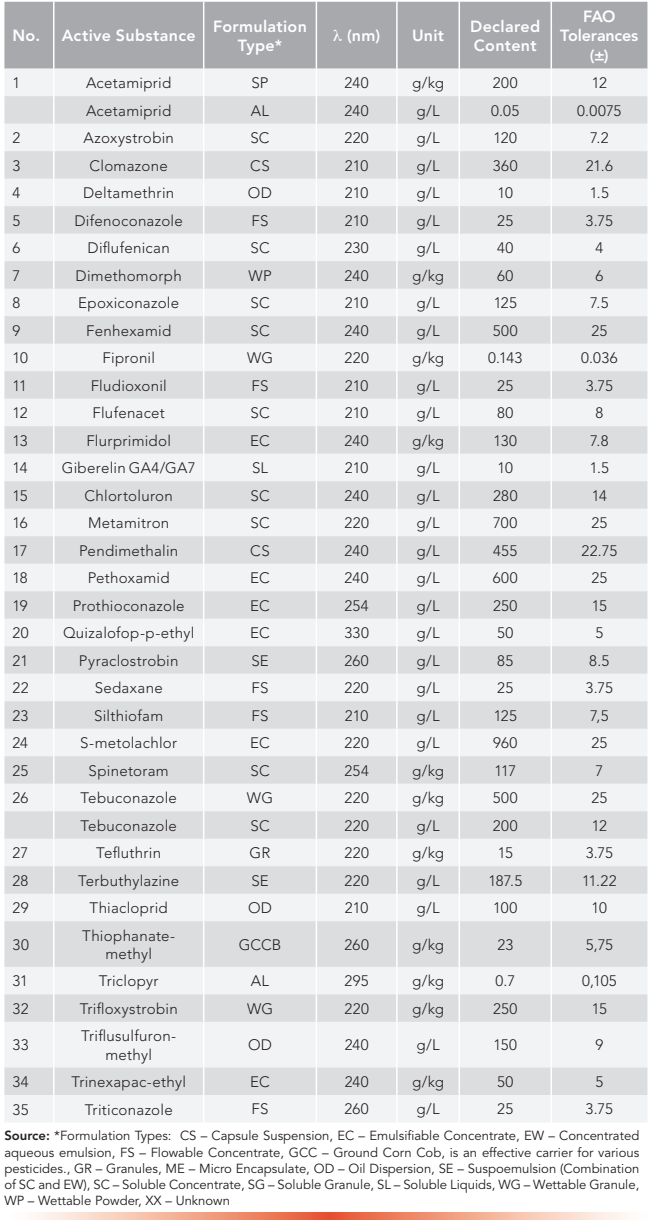
Figures 1 and 2 show example chromatograms for two representative compounds analyzed by the HPLC and UHPLC methods, respectively.

Figure 1: Example chromatogram of Spirotetramat using the HPLC conditions of the multi-analyte method, concentration is 150 g/L. Axis labels are Time (min) for x-axis and Signal (mAU) for y-axis.
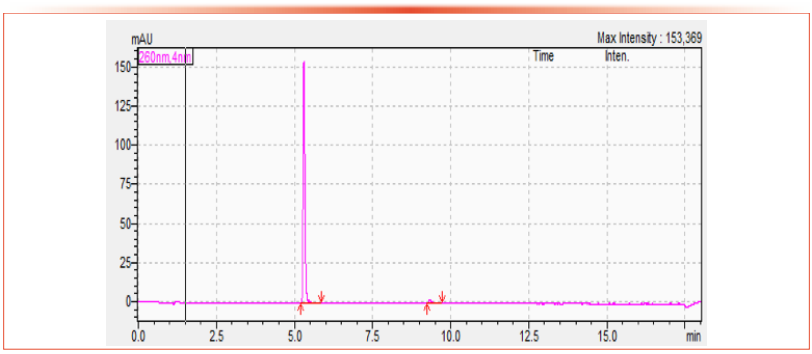

Figure 2: Example chromatogram of Pendimethilin, CS, using the UHPLC conditions of the multi-analyte method, concentration is 455 g/L. Axis labels are Time (min) for x-axis and Signal (mAU) for y-axis.
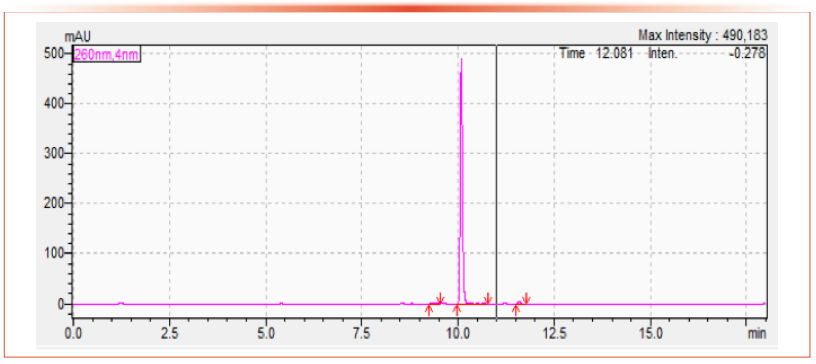
Validation of Technical Materials Using the Multi-Analyte Method
Acceptable linearity of the active ingredient for all the products outlined in Table III and Table IV was demonstrated by using a characterized analytical standard of the same AI using a three-point calibration curve with concentrations at the product concentration and two further points at +50% and -50%. Figure 3 shows a typical calibration curve. Repeatability was calculated via dual preparation and dual injection of each sample. Seven individual active ingredients were used to study the difference in results for Santé et Consommateurs (Directorate General Health and Consumers, European Commission, or SANCO) guideline validation parameters (3) using both the registration method for the technical material and the multi-analyte method described herein. The validation includes linearity, precision, accuracy, and specificity or selectivity. The active ingredients examined during this comparison were chlorantraniliprole, azimsulfuron, bifenthrin, chlorsulfuron, cyantraniliprole, flutriafol, indoxacarb, and triflusulfuron.

Figure 3: Typical calibration curve using the HPLC conditions of the multi-analyte method for the Spirotetramat standard. Axis labels are Concentration (×102) for x-axis and Area (×106) for y-axis.
To prove equivalency, a seven-point calibration curve was determined for all seven active ingredients. As can be seen in Table V, the correlation coefficient for each active using both the registration method and the multi-analyte method was 0.99 or greater. Both method and instrument precision were tested in these experiments. Eight individual weighings of one sample of each active ingredient were measured to determine the method precision. One sample injected eight individual times defined instrument precision. Results of this can also be found in Table V.

Table V: A comparison of linearity and accuracy equivalency data for eight active ingredients by individual GLP enforcement and the multi-analyte method.
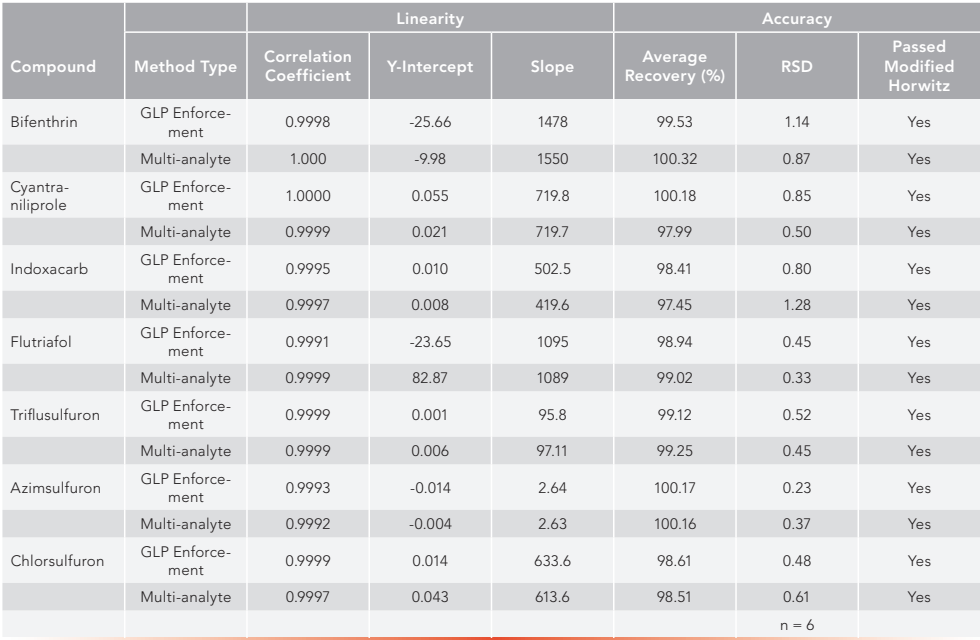
Accuracy equivalency was established by preparing duplicate samples at three separate concentrations. These concentrations were set at 75, 100, and 125% of the GLP sample solution concentration in the enforcement method. Average weight percent results, which ranged from 98.5 to 100.3 with relative standard deviations (RSD) for n=6 ranging from 0.23 to 1.28, all passed the modified Horowitz limit. The data can be found in Table VI.

Table VI: A comparison of precision equivalency data for eight active ingredients by individual GLP enforcement and the multi-analyte method.
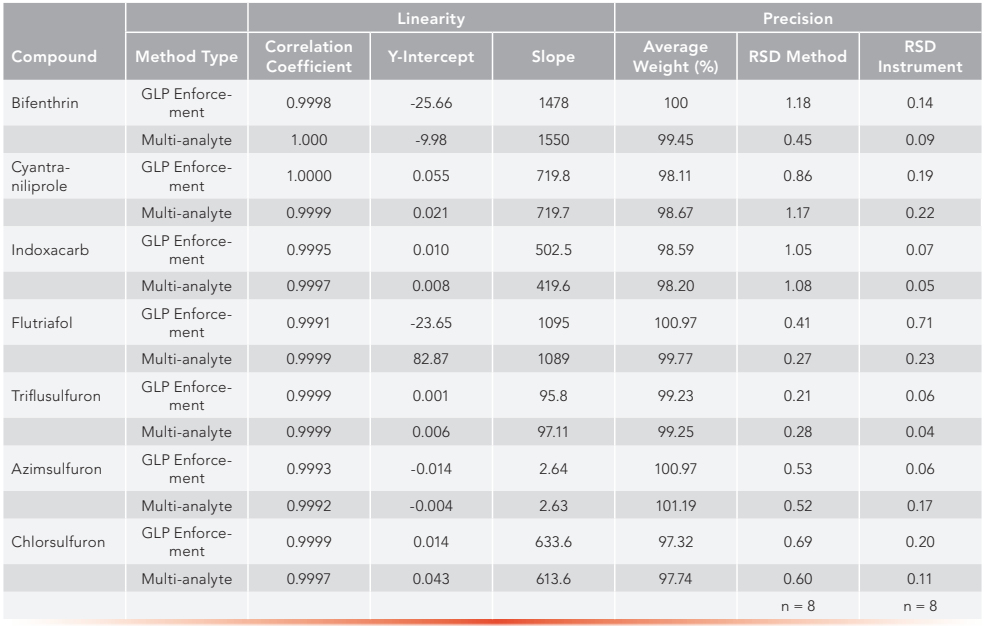
A qualitative analysis for potential impurity interferences for all samples was conducted to determine method specificity. Ideally, an impurity exhibits no interference, and acceptable interference is less than 3% of the overall sample composition. No interferences were seen in either method for the seven active ingredients.
Conclusion
Although the multi-analyte method is not the registered method, it does provide a very useful screening tool for laboratories that see a variety of agricultural product samples. It eliminates the need for a wide variety of columns and can accurately measure the active ingredient content in the formulated product, thus determining whether the material meets the registered manufacturer’s specifications.
The multi-analyte method is evergreen in terms of its future use, and the addition of actives and products are possible if the following criteria are met:
- An unequivocal identity confirmation; preferably mass spectrometry
- Injection repeatability data for standards – RSD should be ≤ 1%
- Linearity should be demonstrated between 50 and 150 mg/kg
- The repeatability for sample analysis should be less than 5%—typically three subsamples of the product and two injections of each sub-sample.
A proficiency test or collaborative trail data would be beneficial to support the use of this methodology for any additional agricultural compounds.
Acknowledgment
The authors would like to express their appreciation to the staff in their laboratories involved in this work.
References
(1) Goldman, L. R. Managing Pesticide Chronic Health Risks. J. Agromedicine 2007, 12 (1), 67–75. DOI: 10.1300/J096v12n01_07
(2) World Health Organization, Fact Sheets, Pesticide Residues in Food, September 15th, 2022.
(3) European Commission, Directorate-General Health and Consumer Protection, SANCO/3030/99 rev 5, March 22, 2019. Technical Material and Preparations: Guidance for generating and reporting methods for analysis in support of pre- and post-registration data requirements for Annex II (part A, Section 4) and Annex III (part A Section 5) of Directive 91/414.
About the Authors
Jim Garvey is with the Food Chemistry Department of Agriculture at the Food and The Marine, Backweston, Laboratory Campus, in Celbridge, Ireland. Olga Nováková is with the Central Institute for Supervising and Testing in Agriculture at the National Reference Laboratory, Department of Testing Plant Protection Products, in Brno, Czech Republic. Olivier Pigeon is with the Walloon Agricultural Research Centre, Building Rachel Carson, in Gembloux, Belgium. Mary Ellen McNally is with FMC Corporation in Newark, Delaware. Direct correspondence to: Mary-Ellen.McNally@fmc.com










Altering Capillary Gas Chromatography Systems Using Silicon Pneumatic Microvalves
May 5th 2025Many multi-column gas chromatography systems use two-position multi-port switching valves, which can suffer from delays in valve switching. Shimadzu researchers aimed to create a new sampling and switching module for these systems.


Analytical Challenges in Measuring Migration from Food Contact Materials
November 2nd 2015Food contact materials contain low molecular weight additives and processing aids which can migrate into foods leading to trace levels of contamination. Food safety is ensured through regulations, comprising compositional controls and migration limits, which present a significant analytical challenge to the food industry to ensure compliance and demonstrate due diligence. Of the various analytical approaches, LC-MS/MS has proved to be an essential tool in monitoring migration of target compounds into foods, and more sophisticated approaches such as LC-high resolution MS (Orbitrap) are being increasingly used for untargeted analysis to monitor non-intentionally added substances. This podcast will provide an overview to this area, illustrated with various applications showing current approaches being employed.


Troubleshooting Everywhere! An Assortment of Topics from Pittcon 2025
April 5th 2025In this installment of “LC Troubleshooting,” Dwight Stoll touches on highlights from Pittcon 2025 talks, as well as troubleshooting advice distilled from a lifetime of work in separation science by LCGC Award winner Christopher Pohl.

Study Examines Impact of Zwitterionic Liquid Structures on Volatile Carboxylic Acid Separation in GC
March 28th 2025Iowa State University researchers evaluated imidazolium-based ZILs with sulfonate and triflimide anions to understand the influence of ZILs’ chemical structures on polar analyte separation.
 Headquarters complex in Washington, DC. | Image Credit: © Tada Images - stock.adobe.com")

