Reaching New Limits for Operational Dynamic Range in Proteomics with a Novel UHR-TOF Design
Bruker Daltonics Inc.
Complex proteomics sample identification is among the most challenging tasks of modern analysis. Identifying, quantifying, and characterizing proteins as comprehensively as possible from low-amount highly-complex samples requires the utmost speed, dynamic range, and sensitivity. The new maXis impact offers a no-compromise solution delivering full resolution at full sensitivity and mass accuracy. It is capable of high sensitivity acquisitions at rates up to 50 Hz in MS and MS-MS with excellent spectral quality obtained under dynamic control of MS-MS acquisition speed. Together with state-of-the art data evaluation capabilities from Bruker's ProteinScape software suite, this package proves to be a powerful tool to address the most challenging proteomic analyses. Here we will demonstrate the performance of the maxis impact using a quantified standard of complex proteins — the Protein Dynamic Standard set from Sigma (UPS-2).
Experimental
The Protein Dynamic Standard Set from Sigma (UPS-2) contains 48 proteins covering five orders of magnitude in concentration. Each concentration level is represented by eight proteins with various pI, molecular weight, and hydrophobicity.


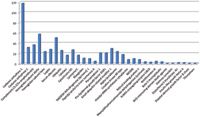
Figure 1: Proteins identified after Mascot search with the corresponding amount on-column and the number of unique peptides per protein.
240 ng of an UPS-2 digest, corresponding to on-column protein amounts ranging from 11 amol to 1.1 pmol have been injected for LC–MS-MS analysis. The sample was applied on a Dionex Ultimate 3000 nanoRSLC system. Separation was performed with a 120 min water/ acetonitrile gradient on a Dionex PepMap C18 column (250 mm × 75 μm). Database search was performed using MASCOT against a dedicated database. Carbamidomethylation was considered as fixed modification whereas methionine oxidation and N-Term protein acetylation were considered as variable modifications. All results were stored and further analyzed using Bruker's ProteinScape 2.1.


Figure 2: Display of the ProteinScape result table , showing the eight most abundant (1.1 pmol on column) and the eight least abundant proteins, covering four concentration levels). Mass accuracy stability is shown by the High Resolution XIC extracted ion chromatogram with a 0.001 Da filter. Such high-selectivity XIC traces enable accurate quantitation.
Results
The 32 most abundant proteins of the USP-2 Mix have been identified over the Mascot threshold (p < 0.05) (Figure 2), thus fully covering the first four concentration levels of the UPS-2 (Figure 1). Such results could be obtained thanks to the system ability to perform high-speed MS-MS acquisition at high sensitivity.


Figure 3: High MS-MS accuracy and spectral quality is key to reliable assignment: 28% of the fragmented peptides have been assigned. Here we show typical MS-MS spectra for two concentrations. Y ions are in blue, B ions in red. MS-MS tolerance is 0.01 Da.
Conclusions
The Maxis Impact combines several technical features to offer an industry first — a benchtop ESIq-TOF which simultaneously delivers maximum resolution (~40,000) at full sensitivity whilst maintaining full mass accuracy. This ensures that low abundance peptides can be detected in the presence of high abundance peptides. It is this unique performance capability which ensures reliable identification of minority proteins in complex mixtures. Combining the performance power of the maxis impact with the evaluation capabilities of ProteinScape addresses one of the most challenging biological sample analysis tasks; the detection, identification, and quantitation of proteins in complex samples.

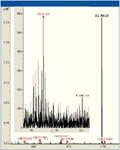
Figure 4: Experimental data have shown the system capability to detect and fragment (red dots are selected peptides) low abundant peptides in the presence of co-eluting abundant peptides. This results from both smart parent selection procedures and a no-compromise technical design which removes the need for artificial dynamic range compensation.
Bruker Daltonics Inc.
Billerica, MA
tel. (978) 663-3660, fax (978) 667-5993
Website: www.bruker.com/maXis
For research use only. Not for use in diagnostic procedures.
















SEC-MALS of Antibody Therapeutics—A Robust Method for In-Depth Sample Characterization
June 1st 2022Monoclonal antibodies (mAbs) are effective therapeutics for cancers, auto-immune diseases, viral infections, and other diseases. Recent developments in antibody therapeutics aim to add more specific binding regions (bi- and multi-specificity) to increase their effectiveness and/or to downsize the molecule to the specific binding regions (for example, scFv or Fab fragment) to achieve better penetration of the tissue. As the molecule gets more complex, the possible high and low molecular weight (H/LMW) impurities become more complex, too. In order to accurately analyze the various species, more advanced detection than ultraviolet (UV) is required to characterize a mAb sample.



