The Perfect Method, Part III: Adjusting Retention
LCGC North America
Faster isn't always better
This is the third installment in a series on method development for liquid chromatography (LC). The focus of the series is on developing new reversed-phase methods in a manner that makes for more reliable methods, while at the same time identifying key areas where problems can occur. In the first section (1), we looked at goal setting, and the second (2) considered the selection of starting conditions. This month's installment of "LC Troubleshooting" will focus on selection of mobile phase conditions that will give reasonable retention of our sample compounds.



Last month, we concluded that a good starting place for most methods was to use a 150 mm × 4.6-mm, 5-μm particle column or a 100 mm × 4.6 mm, 3-μm column for most sample types. This configuration gives enough theoretical plates (N ≈ 10,000 for real samples) to separate most sample types and can be run at 1–2 mL/min for fast method development runs. A mobile phase of low-pH buffer (for example, 25 mM phosphate at pH 2.5) blended with acetonitrile, or optionally methanol, was the mobile phase of choice for UV detection unless you have information to suggest otherwise. For LC–mass spectrometry (MS) applications, a 50 × 2.1-mm, 3-μm particle column operated at 0.2–0.5 mL/min is typical, and 0.1% formic acid is used instead of phosphate buffer. The column temperature is controlled, generally at 30 °C or 35 °C as a starting point.


John W. Dolan
Our Guide
We'll be using equation 1 as our guide through the method development process.

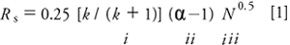
where Rs is the resolution, k is the retention factor, α is the separation factor, and N is the column plate number. Recall that the retention factor (sometimes called the capacity factor, k') is

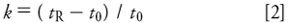
where tR is the retention time and t0 is the column dead time (sometimes abbreviated tm). The column dead time is the retention of an unretained sample component, usually determined by the upslope of the "garbage" or "solvent" peak at the beginning of the chromatogram. We are concerned only about retention (part i of equation 1) this month. In future installments, we'll consider parts ii and iii.
Selecting a Target k-Value
To get the "best" chromatography, we strive for 2 <k < 10, but this often is not possible, so 1 < k < 20 generally is acceptable. When k is in one of these ranges, we'll usually get the best separation, but this is not guaranteed. One of the important reasons we would like k > 2 is that resolution is less susceptible to small fluctuations in mobile phase concentration. This is illustrated in Figure 1 and Table I.

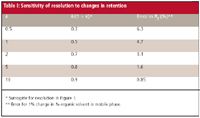
Table I: Sensitivity of resolution to changes in retention
In Figure 1, the influence of retention (term i, equation 1) is plotted against resolution. If k is infinity, term i approaches 1.0, so this is shown as the maximum possible resolution by the dashed line in Figure 1. I like to think of the influence of k on Rs in three ways.
First, let's look at getting the maximum power or leverage out of k as a variable to obtain resolution. If 2 < k < 10, we have achieved 70–90% of the possible resolution by adjusting k (see Figure 1 and column 2 of Table I). This is a pretty good return on our investment in this variable. Increased retention to k ≈ 20 will gain only an additional 5% in resolution, but at a cost of twice the run time and result in broader peaks — not a good tradeoff in my opinion. On the other hand, if k ≈ 1, we're only at 50% of the maximum resolution, so we are not taking full advantage of this variable. For k < 1, resolution drops off rapidly. Thus, we can see that the 2 < k < 10 or 1 < k < 20 guidelines make sense from the standpoint of taking advantage of the power of k to achieve resolution.

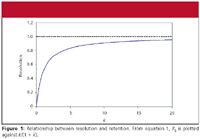
Figure 1
Second, we can consider the susceptibility of a method to small errors in mobile phase composition. For example, if we use a 1% error in mobile phase organic for comparison, we can see that larger k-values are less susceptible to changes in resolution for small changes in percent organic (see Figure 1 and column 3 of Table I). That is, when compared to the error at k ≈ 10 (0.85%), k ≈ 2 has about three times the error and k ≈ 0.5 is more than seven times the error, with a 6.3% change in resolution for a 1% change in organic solvent. So we can see that methods that generate small k-values will be more susceptible to changes in resolution when small changes in mobile phase composition are made, either as a result of normal variation or due to an instrument or operator error.
Finally, real samples almost always generate a large peak at t0, often called the solvent front or garbage peak, due to unretained materials in the sample. In LC–MS, although a peak at t0 usually is absent, a general region of ion suppression exists near t0. In both cases, quantification of analyte peaks can be compromised due to unretained compounds in most samples.
All of these influences support the goal of having 2 < k < 10 for our samples, or if this is not possible, 1 < k < 20. One last way to look at these recommendations is to consider them in light of potential method problems. I have a friend who claims that if every problem LC method were adjusted so that k for the first peak was at least 1, 50% of the problems would go away. I think this is a bit of an oversimplification, but the basic premise is valid. We tend to want to make our method run times short, so peaks get pushed up into the k < 1 region, where there will be more problems with method variation and more likelihood of interference with unretained materials.
What About Run Time?
There is no denying that for most applications, shorter run times are desirable. This is some of the motivation for the current emphasis on sub-2-μm particle columns operated at pressures greater than 6000 psi (400 bar). However, in any application it seems like we will have to trade run time for an increase in k-values. That is, if we have to increase k to get it within the 2 < k < 10 target range, the run will be longer. Contrary to popular opinion, this is not necessarily the case. Consider the case in which the current method gives k = 0.5 for the first peak and we adjust the conditions so that k = 2 for the first peak. For a 150 mm × 4.6 mm column operated at 1 mL/min, t0 ≈ 1.5 min, so we can rearrange equation 2 to solve for tR and figure out the retention time in both cases. For k = 0.5, tR = 2.6 min and for k = 2, tR = 4.5 min, so the run time nearly doubles. However, if we are willing to increase the flow rate, we can gain back some or all of this time. For example, most of us run conventional methods with pressures in the 2000–2500 psi region, yet the upper pressure limit for most traditional LC systems is 6000 psi. You can see that doubling the flow rate will reduce the retention time proportionally, but according to equation 2, a change in flow rate has no effect on the retention factor, because both tR and t0 change in proportion to flow rate. Yes, doubling the flow rate will reduce the column plate number and, thus, resolution, at least in theory. But from a practical standpoint, most methods will not have a noticeable reduction in resolution by a doubling of the flow rate for columns packed with 3- or 5-μm columns. For example, my calculations for a 150 mm × 4.6 mm column packed with 5-μm particles show that a peak pair with k ≈ 2 and Rs = 1.7 will degrade to Rs = 1.6 when the flow is changed from 1 to 2 mL/min. And this is with a well behaved system — most of our methods are not as sensitive to a twofold change in flow rate. So for this example, we have a win-win situation — k is increased to give better chromatography and fewer method problems, yet retention time does not increase. Of course, this assumes that the peak spacing does not degrade with a change in k, which might or might not be true, as we will see in next month's discussion.
Getting k Right
Ok, now we have a target range for k and justification for it, how do we achieve the desired result? One time-honored approach is to start with a strong mobile phase and decrease the mobile phase strength in steps until the desired retention is observed. For example, make a run at 100% acetonitrile, 90%, 80%, and so forth. Then when you are close to the desired result, make small changes to finetune the separation. This technique works well, and when I worked in an application laboratory for one of the instrument companies, it was the standard procedure.
There is a simpler way. If we make a plot of log(k) versus percent organic (%B) in the mobile phase, we will see a graph similar to that of Figure 2. One of the most striking observations about this graph is that it is linear, so it can be described as:

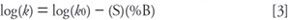
where k0 is the (extrapolated) value of k at 0% B (100% water or buffer) and S is the slope of the plot. Armed with this relationship, we need only two experimental points to make the plot, not a whole series of 10% steps. This means that once we have made two experimental runs, such as 70% and 50% B in this case, we can predict the k-value of our sample compound under any other mobile phase concentration. For this example, it is trivial to determine that k = 1 at 81% B and k = 20 at 48% B, so I should first look within the 48–81% B region for the best k-values, and hopefully the best separation.
As we'll see in next month's "LC Troubleshooting," plots similar to Figure 2 for samples of similar molecular weight will have similar, but not necessarily identical, slopes, or S-values. So even without making experimental plots, we can make a generalization of The Rule of Three, which states that a 10% change in mobile phase organic concentration will change k by a factor of about three. This is not a hard-and-fast rule — it can be 2.5 or 4 for some compounds, and applies for compounds of molecular weights less than ≈ 1000 Da, but it gives us a nice guideline. For example, in the earlier case of a change in k from 0.5 to 2, without any experiments I can guess that the change will require a reduction in mobile phase organic solvent concentration of 10–15%.


Figure 2
Conclusions
We've seen that, from a method development standpoint, it is desirable to adjust retention for that 1 < k < 20, or even better 2 < k < 10. This gives us sample retention times that will give more robust methods in terms of sensitivity to small changes in mobile phase composition. From a troubleshooting standpoint, we can understand that when k < 1 for most methods, besides excessive sensitivity to mobile phase composition, there is more likelihood of quantification problems due to interferences at t0.
The regular behavior of retention and mobile phase organic concentration in reversed-phase LC, as illustrated in Figure 2, gives us a tool to use to more quickly find experimental conditions that will give us retention in the desired range. You might have noticed, however, that a change in %B to change k often results in a change in relative retention, or peak spacing, as well. This can be a problem that can create problems when we try to increase k to move peaks away from t0, but, as we'll see in next month's installment of "LC Troubleshooting," we can use such changes in selectivity to our advantage so as to finetune a separation with very little extra work.
References
(1) J.W. Dolan, LCGC 25(6), 546–552 (2007).
(2) J.W. Dolan, LCGC 25(7), 632-638 (2007).
John W. Dolan "LC Troubleshooting" Editor John W. Dolan is Vice-President of LC Resources, Walnut Creek, California; and a member of LCGC's editorial advisory board. Direct correspondence about this column to "LC Troubleshooting," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail John.Dolan@LCResources.com. For an ongoing discussion of LC trouble-shooting with John Dolan and other chromatographers, visit the Chromatography Forum discussion group at http://www.chromforum.com.




















Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.


