Glossary of Terms Related to Chromatographic Method Validation
LCGC North America
One issue that has become clear to us throughout courses, workshops, seminars, and various talks on the subject of method validation, is that while many people talk the language, sometimes the individual method validation terms mean different things to different people. While the actual protocols, or experimental details used to measure or evaluate method validation can vary, it's a good idea to have a common understanding of the underlying terms. In this month's installment of "Validation Viewpoint," we have compiled glossary of method validation terms as they pertain to chromatography.
Acceptance Criteria
Numerical limits, ranges, or other criteria to which test results for a drug substance, product or other sample should conform to be considered acceptable for its intended use.



Accuracy
The closeness of test results to the true value.
Active Pharmaceutical Ingredient (API or Drug Substance)
Any substance or mixture of substances intended to be used in the manufacture of a drug (medicinal) product that, when used in the production of a drug, becomes an active ingredient of the drug product.
Analytical Instrument Qualification (AIQ)
The process of ensuring that an instrument is suitable for its intended application. AIQ is just one component of data quality that also includes software and analytical method validation, system suitability tests, and quality control tests.
Analytical Performance Characteristics
Parameters assessed during method validation, including accuracy, precision, linearity (range), limit of quantitation or detection, and robustness. Actual parameters assessed depend upon the type of method and its intended use.
Analysis of Variance (ANOVA)
A statistical test that measures the difference between the means of groups of data. Sometimes called an F-test, ANOVA is closely related to the t-test. The major difference is that where the t-test measures the difference between the means of two groups, an ANOVA tests the difference between the means of two or more groups.
Assay (Content or Potency)
An exact result that allows an accurate assessment on the content or potency of the analyte in a formulation.
Assignable Cause
The laboratory investigation following an out-of specification (OOS) result that determines the suspect result is due to a known cause such as operator or instrument error.
Asymmetry
Factor that describes the shape of a chromatographic peak. Theory describes Gaussian symmetrical peak shapes. Peak asymmetry is measured as the ratio of the distance between the peak apex and the back side of the peak to the distance between the front side of the peak. A value > 1 is a tailing peak, while a value < 1 is a fronting peak.


Figure 1
Audit
A documented independent review performed on a periodic basis to verify compliance with a quality system.
Batch
A quantity of material produced during one manufacturing cycle using the same specification.
Biologic
A virus, therapeutic serum, toxin, antitoxin, or related substance used for human treatment or disease prevention.
Biotechnology
The use of living organisms or other biological systems in the manufacture of drugs or other products or for environmental management, for example, waste recycling.
Biological Matrix
Samples of biological origin, typically blood, serum, plasma, urine, feces, saliva, sputum, and tissues.
Blank
A sample that does not include the analytes of interest, used to assess the specificity of a method. Examples include mobile phase, diluent, or procedural blanks.
cGMP
Current Good Manufacturing Practice. See Title 21, Code of Federal Regulations, Section 210, 211 and 212.
Calibration
Ensures that the instrument response correlates with the response of the standard or reference material. Calibration should be carried out by documented written and approved procedures, using traceable certified standards.
Certification
A documented statement, or written guarantee by qualified individuals that an instrument, computer, test or system complies with specified requirements.

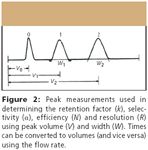
Figure 2
Change Control
A monitoring system of managing and implementing changes that may affect the status of a validated process. Change control is a way to determine the need for corrective actions that might be necessary to correct or redesign systems or, for example, upgrading software while maintaining a validated state.
Coefficient of Determination
The square of the correlation coefficient.
Coefficient of Variation (CV)
The sample standard deviation divided by the sample average, divided by 100. Sometimes called relative precision, or relative standard deviation.
Confidence Interval–Limits
Usually expressed as a percentage (for example, 95%) referring to the range of values around an observed value that will include the expected value.
Confidence Level
Usually expressed as a percentage (for example, 95%) referring to the probability of precision measurements. A 95% confidence level means a 95-in-100 chance of being correct or a 5-in-100 chance of being wrong in predicting that the precision falls into a specified range.
Corrective Action
Action to eliminate the cause of a detected nonconformity to prevent recurrence.
Conformance to Specifications
The sample, when tested according to a documented analytical procedure, satisfies the listed acceptance criteria.

Figure 3
Correlation Coefficient (r)
Degree of correlation between two variables, ranges from -1 to +1. A +1 value indicates a perfect correlation with both values increasing; a value of -1 also indicates a perfect correlation with one value is increasing while the other is decreasing. A zero r value indicates no correlation.
Dead Volume
The volume (Vd) of the chromatographic system not including the column packing. Includes the column interstitial volume, and extra column volume contributed by the injector, detector, tubing and connections. Determined by injecting an inert compound (for example, acetone).
Design Qualification (DQ)
DQ assures that an instrument is designed and produced in a validated environment according to good laboratory practices (GLP), current good manufacturing practices (cGMP), and ISO 9000 standards.
Detection Limit (DL or LOD)
Characteristic of limit tests, the DL is defined as the lowest concentration of an analyte in a sample that can be detected, not quantitated. It is a limit test that specifies whether or not an analyte is above or below a certain value.
Documentation
The organized collection of written or electronic records that describe the structure, purpose, operation, maintenance, and data requirements for systems, instruments, or tests. Includes manuals, procedures, specifications, operating records, final reports, data, and so forth.
Efficiency
The number of theoretical plates (N) in a chromatographic separation. There are several ways to calculate N, one common way is: N = 16(tR - t0)2 where tR is the analyte retention time, and t0 is the retention time of an unretained peak. See also HETP.
Error
Any deviation of the observed value from the true value.
External Standard
The analyte itself, in high chemical purity, which is not added to the actual samples, but is used to generate an external standard calibration plot used to quantitate the analyte in a sample matrix. In order to successfully utilize the external standard method, it is demonstrated, in a reproducible manner, the recovery efficiency of the analyte in the actual sample matrix. Recovery is evaluated by spiking a placebo of the sample matrix with the external standard and showing how much has been recovered. It can then be assumed that the recovery of the same analyte in an actual sample will have the same recovery efficiency.
F Test
A variance ratio test that describes if two independent estimates of variance can reasonably be accepted as being two estimates of the variance of a single, normally distributed sample.
Good Laboratory Practices (GLP)
The organizational process and the conditions under which laboratory studies are planned, performed, monitored, recorded, and reported. See, for example, 21 Code of Federal Regulations (CFR) part 58.
Gradient Elution
Increasing mobile phase strength versus time in a chromatographic run. Gradients can be continuous or step-wise.
HETP
Height equivalent to a theoretical plate, a measure of a chromatographic column's efficiency (N). HETP = L/N, where L is the column length, and N is the number of theoretical plates.
Identification
Ensuring the identity of an analyte.
Impurity
Any component present in the intermediate or API that is not the desired entity.
Impurity Profile
A description of the identified and unidentified impurities present in an API.
Installation Qualification (IQ)
IQ assures that all of the activities associated with properly installing the instrument (new, preowned, or existing) at the users' site are documented.
Intermediate
A material produced during steps of the processing of an API that undergoes further molecular change or purification before it becomes an API.
Intermediate Precision (formerly referred to as ruggedness)
Precision results from within-laboratory variations due to random events such as different days, analysts, equipment, and so forth. Experimental design should be employed so that the effects (if any) of the individual variables can be monitored.
Internal Standard
There are any number of ways to do absolute quantitation in demonstrating accuracy and precision in the validation steps. One is to use an internal standard, which is placed into the sample before any extraction or isolation steps are performed. This standard, in known chemical purity, mimics the analyte of interest in efficiency of extraction from the sample matrix, chromatographic–UV–MS performance properties, and it is eluted close to the analyte of interest but distinct from it in UV–MS profiles. A useful internal standard will have about the same recovery efficiency as the analyte itself from the sample matrix. The absolute, ideal internal standard would be an isotopically (nonradioactive) labeled analyte, in known chemical and isotopic purity, commercially available at reasonable costs, which will then have all the same chromatographic–UV–MS properties as the analyte itself. But, because it is resolvable in the mass spectrometer from the analyte itself, it can serve as the perfect internal standard to quantitate recovery and levels of the analyte itself.
Isocratic
Use of a constant-composition mobile phase in chromatography.
Linearity
The ability of the method to elicit test results that are directly, or by a well-defined mathematical transformation, proportional to analyte concentration within a given range.
Linear Regression
A method that determines the best fit line through a collection of data points that represent the paired values of both an independent and dependent variable.
Linear Velocity (u)
The velocity of the mobile phase moving through the column, in centimeters per second. Related to flow rate by the cross-sectional area of the column.
Lot
A collection of units of a single type or composition manufactured under identical conditions that are expected to have the same quality and uniformity within specifications.
Mean
The average of a series of measurements.
Mean or Standard Distribution
The average of the deviations of individual measurements from the average of the group.
Median
For observations arranged in order of magnitude, the median is the value for which an equal number of observations are above it and below.
Method Transfer
Comparison of key validation parameters between two testing sites.
Method Validation
The process by which it is established, through laboratory studies, that the performance characteristics of the method meet the requirements for its intended purpose.
Multiple Regression Analysis
A type of linear regression in which two or more multivariate independent variables are fit to a linear model of one dependent variable.
Nominal Concentration
Theoretical or expected concentration.
Normal or Gaussian Distribution
A sampling of data defined by a mean and a standard deviation that exhibits the frequency of a bell-shaped or Gaussian curve.
Observation
Experimentally derived data.
Operational Qualification (OQ)
OQ testing is done to verify that the instrument or instrument modules operate as intended.
Out of Specification (OOS) Results
All suspect results that fall outside specifications or established acceptance criteria.
Outlier
Data that fails to meet a statistical test for acceptance.
Partial Validation
Validation of the affected performance characteristics of a method or procedure resulting from changes to the methods or test substances. See also: Verification.
Percent Recovery
The observed or assay value divided by the true or theoretical value multiplied by 100.
Performance Qualification (PQ)
PQ testing is performed under the actual running conditions across the anticipated working range. In practice a known method, with known, predetermined specifications is used to verify that all of the modules are performing together to achieve their intended purpose. In practice, OQ and PQ frequently blend together in a holistic approach. For HPLC, the PQ test should use a method with a well-characterized analyte mixture, column, and mobile phase. Actual user PQ tests should incorporate the essence of the system suitability section of the General Chromatography Chapter 621 in the USP in order to show suitability under conditions of actual use.
Precision
The degree of agreement among individual test results when an analytical method is used repeatedly to multiple samplings of a homogeneous sample. See Repeatability, Intermediate Precision, and Reproducibility.
Procedure
A specified way to perform an activity by execution of an approved document intended to produce a result defined by a specification.
Procedural Blank
A sample of known composition (for example, placebo) that does not contain the analyte of interest that is processed, prepared or handled in the same way (procedure) as an unknown sample or standard.
Purity Tests
Analytical procedure that accurately assesses the impurity content of an analyte; for example, related substances test, heavy metals, and residual solvent tests.
Qualification
A subset of the validation process that verifies proper module and system performance prior to the instrument being placed on-line in a regulated environment.
Quality Assurance (QA)
The sum total of the organised arrangements made with the object of ensuring that all APIs are of the quality required for their intended use and that quality systems are maintained.
Quality Control (QC)
Checking or testing that specifications are met.
Quality Control Samples
Samples run to make sure the instrument has been properly calibrated or standardized. Quality control samples are also often used to provide an in-process assurance of the test's performance during use.
Quantitation Limit (QL or LOQ)
The lowest concentration of an analyte in a sample that can be determined (quantitated) with acceptable precision and accuracy under the stated operational conditions of the method.
Range
The interval between the upper and lower levels of analyte (inclusive) that have been demonstrated to be determined with a suitable level of precision, accuracy and linearity using the method as written.
Reanalyze
Repeating the analysis or performing different analyses on the original sample preparation, reference standards or reagents.
Recovery
Extraction efficiency generally reported as a percentage of the known amount of an analyte.
Repeatability
Precision results of the method operating over a short time interval under the same conditions (inter-assay precision). Generally the criteria of concern in USP procedures.
Reproducibility
Precision results of collaborative studies between laboratories.
Resample
To obtain a new sample aliquot from the original test substance source. The sample is used for retesting and should be taken from the same homogeneous material that yielded the out-of-specification (OOS) result.
Resolution (RS)
The separation of two chromatographic peaks that takes into account the retention times and peak widths. Calculated by the equation: RS = 2(tR2 - tR1)/(w± + wB2). A value of 1.0 is considered to be the minimum for a separation to occur, values of 1.5 or better for good quantitation, and values of 2.0 or higher for robustness or disparate levels like those found in impurity profiles.
Retention Factor
A chromatographic parameter (k) that measures the degree of retention. Calculated from the equation: k = (tR-t0)/t0, where tR is the retention time, and t0 is the retention time of an unretained peak.
Retest
Repeating the analytical procedure on a resampled aliquot. See Resample.
Robustness
The capacity of a method to remain unaffected by small, deliberate variations in method parameters; a measure of the reliability of a method. Often used to set the system suitability parameters of a method.
Ruggedness
See Intermediate precision
Signature (signed)
The record of the individual who performed a particular action or review. This record can be initials, full handwritten signature, personal seal, or authenticated and secure electronic signature.
Specification
A list of tests, references to analytical procedures, and appropriate acceptance criteria for the test described.
Specificity
Specificity is the ability to measure accurately and specifically the analyte of interest in the presence of other components that may be expected to be present in the sample matrix.
Stability
Degree or rate of degradation of an analyte in a given matrix under specific conditions over a given time interval.
Stability Indicating Method (SIM)
A validated, quantitative analytical procedure used to accurately, precisely and selectively detect a decrease in the amount of the active pharmaceutical ingredient (API) from potential interferences like degradation products, process impurities, excipients, or other potential impurities.
Standard Additions
First used in trace metals analysis in atomic absorption or emission spectroscopy and for organic analytes; a method of quantitating an analyte in complex sample matrices at low levels. The method relies on first dividing the sample solution into approximate equal fractions or volumes, and then analyzing the first sample unspiked, then each additional fraction spiked with known amounts of the authentic analyte at varying levels (1/2 x, x, 2x, 3x, and so on).
Standard Curve
Relationship between the analytical concentration and the experimental response value (also referred to as a calibration curve).
Stock Solutions
Solutions prepared from reference material used for preparation of working solutions (also referred to as primary stock solutions).
System Suitability
System suitability is the checking of a system to ensure system performance before or during the analysis of unknowns. System suitability tests are an integral part of chromatographic methods, and are used to verify that the resolution and reproducibility of the system are adequate for the analysis to be performed. System suitability tests are based on the concept that the equipment, electronics, analytical operations, and samples constitute an integral system that can be evaluated as a whole. System suitability parameters are established as a direct result of robustness studies.
Tailing
Situation where a normally Gaussian peak has an asymmetry > 1.
Validation
A documented program that provides a high degree of assurance that a specific process, method, or system will consistently produce a result that accomplishes its intended purpose, meeting pre-determined acceptance criteria.
Validation Protocol
A written plan stating how validation will be conducted and defining acceptance criteria.
Verification
An assessment of selected Analytical Performance Characteristics of method validation to generate appropriate relevant data as opposed to repeating the entire validation process. Verification demonstrates that acceptable results utilizing the laboratories' personnel, equipment, and reagents can be obtained.
Working Solution
Stock solution dilution used for the preparation of calibration standards and quality control samples (also referred to as secondary stock solutions).
Don't see your favorite term here? Disagree or want to add to a particular term?
Email me at mswartz@synomicspharma.com and we'll try to include it in a future column.
References
(1) M.E. Swartz and I.S. Krull, Analytical Regulatory and Validation Compliance Primer (Marcel Dekker, Inc., New York, 1997).
(2) L.R. Snyder, J.J. Kirkland, and J.L. Glajch. Practical HPLC Method Development, Second Edition (Wiley-Interscience, New York, 1997).
(3) B.A. Bidlingmeyer. Practical HPLC Methodology and Applications (Wiley-Interscience, New York, 1992).
(4) J.J. Kirschbaum, J. Pharm. Biomed. Anal. 7(7), 813 (1989).
(5) J. Kirschbaum, S. Perlman, J. Joseph, and J. Adamovics, J. Chrom. Sci. 22, 27 (1984).
(6) J.K. Taylor, Anal. Chem. 600A–608A (1983).
(7) T.D. Wilson, J. Pharm. Biomed. Anal. 8(5), 389 (1990).
(8) H.T. Karnes and C. March, J. Pharm. Biomed. Anal. 9(10–12), 911 (1991).
(9) H.T. Karnes, G. Shiu, and V.P. Shah, Pharm. Research 8(4), 421 (1991).
(10) E.L. Inman, J.K. Frischmann, P.J. Jimenez, G.D. Winkel, M.L. Persinger, and B.S. Rutherford, J. Chrom. Sci. 25, 252 (1987).
(11) D.R. Williams, BioPharm, 34 (1987).
(12) A.J. Vanderwielen and E.A. Hardwidge, Pharm. Tech. 66 (1982).
(13) G.P. Carr and J.C. Wahlichs, J. Pharm. Biomed. Anal. 8(8–12), 613 (1990).
(14) A.R. Buick, M.V. Doig, S.C. Jeal, G.S. Land, and R.D. McDowall, J. Pharm. Biomed. Anal. 8(8–12), 629 (1990).
(15) V.P. Shah, K.K. Midha, S. Dighe, I.J. McGilveray, J.P. Skelly, A. Yacobi, T. Layloff, C.T. Viswanathan, C.E. Cook, R.D. McDowall, K.A. Pittman, and S. Spector, J. Pharm. Sci. 81(3), 309 (1992).
(16) V.P. Shah, K.K. Midha, S. Dighe, I.J. McGilveray, J.P. Skelly, A. Yacobi, T. Layloff, C.T. Viswanathan, C.E. Cook, R.D. McDowall, K.A. Pittman, and S. Spector. Pharm. Res. 9(4), 588 (1992).
(17) I.S. Krull, J.R. Mazzeo, and C.M. Selavka, Biomed. Chromatogr. 6, 259 (1992).
(18) M.E. Swartz and I.S. Krull, Pharm. Tech. 104 (1998).
(19) I.S. Krull and M.E. Swartz, LCGC 16(3), 258 (1998).
(20) I.S. Krull and M.E. Swartz, LCGC 16(5), 464 (1998).
(21) B.R. Thomas and S. Ghodbane, J. Liquid Chromatogr. 16, 1983 (1993).
(22) K.D. Altria, J. Chromatogr. 646, 245 (1993); b. K.D. Altria, N.G. Clayton, M. Hart, R.C. Harden, J. Hevizi, J.V. Makwana, and M.J. Portsmouth, Chromatographia 39, 180 (1994).
(23) H. Watzig and C. Dette, J. Chromatogr. 636, 31 (1993); J. Chromatogr. 636, 31 (1993).
(24) B.R. Thomas and S. Ghodbane, J. Liquid Chromatogr. 16(9/10), 1983 (1993).
(25) M.E. Swartz, J. Liquid Chromatogr. 14(5), 923 (1991).
(26) G.M. McLaughlin, J.A. Nolan, J.L. Lindahl, R.H. Palmieri, K.W. Anderson, S.C. Morris, J.A. Morrison, and T.J. Bronzert. J. Liquid Chromatogr. 15(6/7), 961 (1992).
(27) Capillary Electrophoresis Guidebook, Principles, Operations, and Applications, K.D. Altria, Ed. Methods in Molecular Biology, Volume 52, (Humana Press, Clifton, New Jersey, 1996).
(28) Trends in Analytical Chemistry, Volume 18, Numbers 9-10, September/October, 1999. Special Issue: Metrology in Chemistry.
(29) Interpharm Company publications on Method Validation, see website for listing; www.interpharm.com
(30) Pharmaceutical and Biomedical Applications of Liquid Chromatography, C.M. Riley, W.J. Lough, and I.W. Wainer, Eds. (Elsevier Science, Inc., Amsterdam, 1994).
(31) Development and Validation of Analytical Methods, C.M. Riley and T.W. Rosanske, Eds. (Pergamon Press, Elsevier Science, Tarrytown, New York, 1996).
(32) L. Huber, Validation and Qualification in Analytical Laboratories (Interpharm Press, Inc., Buffalo Grove, Illinois, 1999).
(33) J. Flarakos, I.S. Krull, and M.E. Swartz, LCGC 19(3), 304 (2001).
(34) I.S. Krull and M.E. Swartz, LCGC 19(7), 604 (2001).
(35) R.C. Thompson, S. Morris, M.E. Swartz, and I.S. Krull, LCGC 19(11), 1142, (2001).
(36) I.S. Krull, "Adventures in Analytical Chemistry/Biochemistry/Biotechnology," in A Century of Separation Science, H. Issaq, Ed. (Marcel Dekker, Inc., New York, 2001), Chapter 44, pp. 693-709.
(37) V. Grisanti, E.J. Zachowski, I.S. Krull, and M.E. Swartz, LCGC 20(4), 356 (2002).
(38) P. Nader, G. Kremmydiotis, J. Tremblay, G., Levesque, A. Bartlett, I.S. Krull, and M.E. Swartz, LCGC 20(9), 856 (2002).
(39) I.S. Krull, American Laboratory 34(20), 6 (2002).
(40) A.S. Rathore, R.R. Kurumbail, A.M. Lasdun, M.E. Swartz, and I.S. Krull, LCGC 20(11), 1042 (2002).
(41) M.E. Swartz and I.S. Krull, LCGC 21(2), 136 (2003).
(42) J. Orr, I.S. Krull, and M.E. Swartz, LCGC 21(7), 626 (2003).
(43) J. Orr, M.E. Swartz, and I.S. Krull, LCGC 21(12), 1146 (2003).
(44) I.S. Krull and M.E. Swartz, American Laboratory, Guest Editorial, 6 (February 2004).
(45) M.E. Swartz and I.S. Krull, LCGC 22(2), 132–136 (2004).
(46) M.E. Swartz, I.S. Krull, and J. McCabe, LCGC 22(9), 906 (2004).
(47) M. Swartz and I.S. Krull, LCGC 22(6), 536 (2004).
(48) I.S. Krull and M.E. Swartz, LCGC 23(1), 46 (2005).
(49) M.E. Swartz and I.S. Krull, LCGC 23(6), 46, 2005.
(50) M. Swartz and I. Krull, LCGC 23(9), 1100 (2005).
(51) P. Lukulay, J. Morgado, M. Swartz, and I.S. Krull, LCGC 24(2), 150 (2006).
(52) M.E. Swartz and I.S. Krull, LCGC 24(5), 480 (2006).
(53) I.S. Krull and M.E. Swartz, The Pharmaceutical Regulatory Guidance Book, July, 2006, pp. 18-23.
(54) M.E. Swartz and I.S. Krull, LCGC 24(8), 770 (2006).
(55) M.E. Swartz and I.S. Krull, LCGC 25(1), 48-55 (2007).
(56) USP 29, 2006, Chapter 1225, Validation of Analytical Procedures.
(57) USP Chapter 621 Chromatography.
(58) International Conference on Harmonization, Harmonized Tripartite Guideline, Validation of Analytical Procedures, Text and Methodology, Q2(R1), November 2005, See www.ICH.org.
(59) AAPS PharmSciTech 2004, 5(1) Article 22: Analytical Instrument Qualification, (http://www.aapspharmscitech.org).
(60) Pharmacopeial Forum, Analytical Instrument Qualification,31(5), Sept-Oct 2005, p. 1453-1463.
Michael E. Swartz
"Validation Viewpoint" Co-Editor Michael E. Swartz is Research Director at Synomics Pharmaceutical Services, Wareham, Massachusetts, and a member of LCGC's editorial advisory board.


Michæl E. Swartz
Ira S. Krull
"Validation Viewpoint" Co-Editor Ira S. Krull is an Associate Professor of chemistry at Northeastern University, Boston, Massachusetts, and a member of LCGC's editorial advisory board.


Ira S. Krull
The columnists regret that time constraints prevent them from responding to individual reader queries. However, readers are welcome to submit specific questions and problems, which the columnists may address in future columns. Direct correspondence about this column to "Validation Viewpoint," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgcmag.com.








Newsletter
Join the global community of analytical scientists who trust LCGC for insights on the latest techniques, trends, and expert solutions in chromatography.




: An HPLC 2025 Video Interview with Torgny Fornstedt")





Researchers Explore Gas Chromatography to Improve Forensic Ink Analysis
July 25th 2025Ink authentication is often complicated by tampering, aging, and chemical variability. Now, forensic scientists are turning to advanced gas chromatography (GC) techniques to enhance the accuracy and efficiency of ink analysis.






