Different Approaches to Synthesizing Molecularly Imprinted Polymers for Solid-Phase Extraction
LCGC North America
Molecularly imprinted polymers (MIPs) are synthetic polymeric materials that mimic immunosorbents. They are widely used as sorbents for solid-phase extraction (SPE). The most common way to synthesize them is bulk polymerization because of its simplicity and versatility. This produces a hard monolith that has to be ground and sieved to obtain particles in the desired size range. However, the partial loss of the materials as fine dusts; the irregular shape of the particles produced and their wide size distribution, have led to a search for different polymerization methods to offset the drawbacks of the bulk polymerization process.
The analysis of organic contaminants in environmental matrices or pharmaceutical compounds in biological fluids (such as plasma or urine) often requires pretreatment steps because of the low concentration levels of the target analytes and the complexity of the matrices. Solid-phase extraction (SPE) is the most commonly used technique. However, many interfering compounds might be coextracted with the target analytes on conventional sorbents.



To overcome this lack of selectivity highly selective sorbents such as immunosorbents (ISs) (1,2) and molecularly imprinted polymers (MIPs) (3) were developed. They allow extraction, concentration and clean-up in a single step. ISs provide a high degree of molecular recognition because of the high affinity and the high selectivity of the antigen–antibody interactions. Nevertheless, their synthesis involves the production of antibodies which is a time-consuming, tedious and expensive process. An alternative technology that uses MIPs was proposed by chemists; it represents an attractive approach to mimic highly specific antibody binding-sites (4). It involves the copolymerization of functional monomers and cross-linking agents in the presence of an imprint molecule (template), which leads to the formation of a rigid polymer.
The removal of the template results in well-defined cavities within the polymer whose structure and arrangement of functional groups are complementary to the template. However, the template removal is usually uncompleted. This problem is common to all MIP formats; its consequences depend on the final use of the sorbent. In the situation of trace analysis, the template molecules' bleeding gives rise to erroneous results, thus an exhaustive washing of the polymer with a range of solvents should be performed before use to extract the entire amount of the template molecules
The solvents to be used should promote polymer swelling and disrupt template/polymer interactions. Commonly used solvents are methanol or acetonitrile with the addition of acidic or basic modifiers; they need to be optimized for each type of polymer. MIPs have a large number of potential applications but most activities are focused on their use in solid-phase extraction (SPE). Their use as selective sorbents in SPE was first performed by Sellergren and colleagues (5), more than 20 years ago, for the extraction of pentamidine from urine. Since this pioneering work, the number of papers dealing with the application of MIPs in solid-phase extraction (MIP–SPE) is increasing (6,7).
Most of the reported MIPs were prepared by bulk polymerization which results in a monolith that should be ground and sieved to obtain irregular particles. Other polymerization methods can be performed to directly obtain better defined particles such as precipitation, suspension and multi-step swelling. The principle of each method and their relative benefits and limitations are listed in Table I. This article discusses different procedures used for preparing MIPs mainly dedicated to SPE.


Table I: Comparison of the different polymerization methods. Adapted from reference 48 and reprinted with permission
Principle of MIP–SPE
The principle of selective extraction on MIPs is similar to the extraction with ISs. In the off-line mode, the MIP particles are packed into a disposable cartridge between two frits. Figure 1 illustrates the off-line MIP–SPE procedure. After a conditioning step, the sample is percolated through the MIP and a selective washing step allows the elimination of interfering compounds retained by nonspecific interactions. The target analytes are then eluted by the percolation of a solvent able to disrupt the selective interactions between the binding sites and the analytes to recover them. The residue obtained can be analyzed either by liquid chromatography (LC) or gas chromatography (GC). In the on-line coupling, the MIP sorbent is slurry packed into a pre-column which is located in a six-port switching valve (8–20) connected to a LC system.


Figure 1
In the load position, the MIP–SPE is performed and, in the inject position, the MIP precolumn is connected to the analytical column. This allows simultaneous elution and transfer of the retained analytes by the mobile phase of the LC separation. The on-line coupling of SPE with a LC system provides higher enrichment factors compared with the off-line system (21).
The nature of the various solvents involved in a MIP–SPE procedure can be very different from those used in immunoextraction. Because biological reagents are used, ISs are particularly well adapted to the direct percolation of aqueous samples. The washing and the elution steps are achieved with the use of hydro-organic mixtures (7). In return, MIP–SPE procedures consist mainly on the use of organic solvents because MIPs offer the highest selectivity when samples are diluted in the solvent used for their synthesis (22).
As most MIPs are synthesized in nonpolar and aprotic solvents, MIPs seem to be well adapted to the clean-up of complex matrices. The direct extraction of compounds from aqueous matrices on MIPs is more difficult. During the percolation of the aqueous sample, the trace analytes' retention is mainly ensured by nonselective hydrophobic interactions with the polymeric matrix. A careful choice of the washing solvent should be done; it should enhance the development of selective polar interactions between the analytes and the cavities. Thus, the use of a control polymer, a nonimprinted polymer (NIP), in parallel is essential to evaluate the real selectivity of the MIP and the risk of contribution of the nonspecific interactions.
The NIP is a reference polymer obtained by performing the synthesis procedure without the template. Therefore, the retention on this sorbent only results from nonspecific interactions. A better knowledge of retention mechanisms helps for the efficient use of MIP–SPE in aqueous media. It is important to underline that MIPs are not intrinsically selective. It is the MIP–SPE procedure (conditions of percolation, washing and elution) that confers the MIP its selectivity (7). Theoretically, the best results expected when using MIP–SPE correspond to the achievement of a recovery close to 100% on the MIP with no retention on the NIP after percolating real samples containing the target analyte(s).
Results close to this optimal situation were obtained for the selective extraction of triazines from real waters (23), buvicaine and analogue (24) or alfuzosin from plasma (25). In the situation of triazines, the potential of the MIP–SPE was compared with an immunosorbent and a similar improvement of selectivity was obtained. Recently, Chapuis and colleagues reported the use of a MIP prepared by bulk for the clean-up of a basic drug, alfuzosin, from plasma and from soil extract (25). In both instances, high recoveries and high degree of selectivity were obtained. The enhancement of selectivity brought by the MIP for the treatment of soil extract is illustrated in Figure 2. The resulting chromatogram [Figure 2(b)] was compared to the direct injection of the soil extract without MIP clean-up [Figure 2(a)] and to the direct injection of standard solution of alfuzosin [Figure 2(c)]. This application shows the potential of MIP for the sample treatment of various matrices. The obtained chromatograms show that MIP clean-up provides a cleaner baseline and allows an easier determination of the target analyte with high recovery.

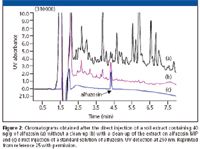
Figure 2
Reagents
The success of the imprinting experiment relies on the careful choice of the reagents: (1) the template molecule, (2) the functional monomers, (3) the cross-linking agent, (4) the polymerization solvent called the porogen and (5) the radical initiator (26). The most common reagents used for MIP synthesis are reported in Table II. A MIP is usually synthesized for an intended analytical use that implies the choice of a given template molecule. The structure and the functionalities of this molecule define the subsequent properties of the binding sites. Criteria to consider when selecting a candidate molecule that will serve as "mould" are cost, availability and the interactions sites.

Table II: Most common reagents used in molecular imprinting
Usually, the amount of template used is high (often 1 mmol), therefore expensive or difficult to synthesize templates can be substituted by a structural analogue to decrease the cost of the material. In noncovalent imprinting, the interactions involved are weak so the candidate template should present multiple functional sites to increase the strength of the template-functional monomers assemblies. The use of a structural analogue (27) or of an isotope (28) as an alternative imprint molecule is known as "dummy imprinting."
The resultant MIP should give rise to imprints that have the ability to bind the target analyte. In this instance, the template leaching will not lead to erroneous results. When a candidate template is selected, the choice of functional monomers able to strongly bond it before polymerization is crucial. The strength of the interactions and the arrangement of the functional groups influence the binding properties of the obtained polymer. In MIP–SPE the most common approach consists on the noncovalent imprinting so the obtained assemblies are mainly based on polar interactions such as hydrogen bonding and ionic interactions.
The functional monomer should bear the required functional groups complementarily to the template. As a general guidance, for basic templates such as templates containing Brönsted basic functional groups or amine bases, acidic monomers such as methacrylic acid (MAA) are preferably chosen. Basic functional monomers such as 2- or 4-vinylpyridine are more suited for templates containing acid groups. However, we note that MAA is by far the most commonly used monomer because it acts as a hydrogen bond and proton donor as well as a hydrogen bond acceptor. The stabilization of the obtained assemblies is one of the key success factors of the imprinting process.
The noncovalent adducts stability is amended by the reasoned choice of the porogen which is the last listed factor but one of the most important. The imprinting efficiency is enhanced when the polymerization reaction is performed in a nonprotic solvent as apolar as possible (the most widely used solvents are acetonitrile, chloroform, dichloromethane and toluene). The use of protic and/or polar solvents, such as water or ethanol, will hardly suppress hydrogen bonding, which is one of the most involved interactions in noncovalent imprinting. The template–monomer assemblies will be less stable and even totally broken and this may lead to the loss of efficiency. The criteria of choice of the reagents discussed above should be taken into account, despite the format in which synthesis will occur.
Bulk Polymerization
Bulk polymerization is by far the most widely used method for the preparation of MIPs for conventional scale owing to its simplicity and versatility. It consists of the synthesis of a block monolith polymer that has to be ground and sieved to obtain particles of an appropriate size range for subsequent use. Usually, these particles are sieved to yield irregular particles with a nominal diameter in the 25–35 μm range. In the last step, particles are sedimentated to remove fines then dried and packed between two frits into either disposable cartridges to be used in an off-line system or into precolumns to be coupled on-line with LC.
Currently, imprinted particles produced by the bulk process are more often used in SPE. MIP particles were applied to the extraction of compounds from environmental samples (water, ground water, soil extracts) (8,23,29–31), food (32–35), biological fluids, such as urine and plasma (36–39), and tissue samples (27,40,41). Recently, some research groups prepared a porous MIP monolith in-situ into a stainless steel column, which is used first as stationary phase. The MIP selectivity towards the template was studied by comparing MIP retention properties to those of the NIP.
Then, the monolithic polymer was pushed out of the column, ground in a mortar and sieved. The particles obtained were packed into disposable cartridges and used as a SPE sorbent. This approach was applied for the extraction of sinomenine from herb and plasma (42). The potential of MIP prepared by bulk for SPE is highlighted in a large number of scientific reports (4,6,7, 43,44).
Although it is easy to prepare, bulk polymerization is time consuming, labor intensive and wasteful because only 30–40% of the ground polymer is usually recovered as useable material (45). Moreover, the particles produced are far from ideal because of their irregular shape and wide size range. They packed poorly in columns and created large void volumes, which may be a problem when the MIP is coupled on-line to LC. These significant drawbacks limited the suitability of MIPs especially for HPLC separations and binding assays but they don't limit their use in SPE, especially when MIP–SPE is performed in the off-line mode.
Several attempts were investigated to overcome the drawbacks of the bulk process. Producing monodispersed MIP beads by applying new methods, such as suspension polymerization, multi-step swelling polymerization and precipitation polymerization, were reported. The newest approach consists of the in-situ polymerization of monoliths into capillaries. Owing to the increasing interest in nanotechnologies, considerable progress has been made to adapt MIPs to miniaturized separation devices.
Precipitation Polymerization
In a precipitation process, the polymerization occurs in the presence of a larger amount of porogen than that typically used in the bulk method (2–10 times higher). This more dilute polymerization mixture is initially homogeneous. However, as the polymerization proceeds, the growing polymer chains become insoluble in the liquid phase so they precipitate and the solution becomes heterogeneous. Micro- and nanospheres can be generated when accurate control of the parameters governing the precipitation polymerization is completed (that is, polymerization temperature, cross-linker and so on).
Generally, beads have diameters ranging from 0.1–1.0 μm. The diameter dispersion is usually narrowed by the centrifugation. The reaction yield is usually high (>85%) and recognition properties and loading capacities may be higher than those obtained by the bulk process (45,46). However, the presence of the print molecule affects the polymer morphology (specific area and pore size distribution) and even hinders spherical beads being obtained (46–48). It was reported that the particles obtained are colloidal and slightly irregular in shape (48).
Precipitation polymerization was used for the production of MIPs for different phenylurea herbicides such as fenuron (16), linuron, isoproturon (46) and propazine (49). The synthesized imprinted beads were used as SPE sorbents for the clean-up of plant extracts in the off-line mode. Tamayo and colleagues have prepared a fenuron MIP with homogenous binding sites because the degree of homogeneity was close to that reported for MIPs prepared by the covalent approach (16).
The determination of fenuron in plant extracts is made possible with a MIP–SPE clean-up procedure (Figure 3). These results showed that precipitation is a powerful method that enables the preparation of MIPs particles with improved characteristics (size distribution, shape, imprinting efficiency, mass transfer kinetic). It was reported that the presence of the template prevents spherical beads being obtained but this is not a limitation for SPE applications (46).


Figure 3
The precipitation polymerization may be one of the easiest methods for the preparation of MIPs particles because crushing and sieving steps are avoided and higher yields of reaction are obtained.
Suspension Polymerization
Suspension polymerization is a heterogeneous polymerization method for the production of spherical beads in a broad size range starting at a few micrometres and reaching up to millimetres in size (50). In this process, the organic-based polymerization mixture is suspended as droplets into an excess of a continuous dispersion phase by an agitation method (for example, stirring). The dispersant medium can be either water or perfluorocarbon fluids (51,52). We note that compared with the bulk process, the suspension polymerization is performed at a higher molar concentration of template and monomers to compensate the partial loss of the reagents in the dispersant phase which, therefore, are not involved in the polymerization (12). The droplets suspension is not a stable system, thus a stabilizer (suspending agent) is added to the dispersion medium to prevent coalescence of the droplets and the polymerizing chains colliding during the process. The molecules of stabilizer adsorb at the surface of the droplets and form a thin layer that plays the role of a steric barrier preventing the coalescence (53). Polymerization proceeds in the droplets phase by a free radical mechanism, each droplet acts like a mini-bulk reactor.
The solubility of both of the dispersed droplets phase and the resultant polymer in the dispersion medium are low. The produced polymer is soluble in the polymerization mixture so a gel is formed within the droplet at a low conversion rate and this leads to harder spherical beads at a high conversion rate. Polymerization is usually achieved in a simple one-step process and within a short time compared with the bulk process (about 3 h). Once the reaction is completed, the polymer beads are thoroughly washed to remove unreacted components. Drying following the final wash with the porogen preserves the well-defined porous structure of the beads.
The final size of the formed beads is controlled by the ratio of the dispersed and the continuous phases, the stirring rate, the type and the level of the suspension stabilizer etc. The suspension polymerization in water is relatively easy to perform and a large number of suitable recipes can be found in the literature (54,55). However, in the instance of MIPs the aqueous phase may interfere with the interactions established between the template and the monomers. Mayes and colleagues (51) reported the suspension polymerization in perfluorocarbon fluids, which are preferred to water because they are inert, stable and largely immiscible with organic compounds and water. They did not disrupt the interactions between the functional monomers and the template.
Lai and colleagues prepared two types of trimethoprim (TMP) MIPs by bulk and by suspension polymerization (12). Both MIPs were used as sorbents for the selective extraction of TMP from human urine and pharmaceutical tablets. They complete separating and enriching of TMP from the human urine and the pharmaceutical tablet successfully, but the MIP particles based on bulk polymerization were more selective. A recent work reported the direct synthesis of MIP beads in an SPE cartridge by suspension polymerization in a perfluorocarbon fluid (56). The selectivity of the resultant sorbents was tested by radio-ligand binding assays. This approach seems interesting especially when the synthesis of a large number of polymers is required.
The suspension method is reliable and attractive because it produces polymer beads with a considerable yield and improved chromatographic characteristics; particularly good flow-through properties with low back pressure. Although the stirring rate in the reactor provides some control over the average size of polymer beads; this turbulence-based method affords beads with a broad sizes distribution that affects both the column flow through and the efficiency.
The applications of MIP prepared by suspension polymerization are mainly focused on their use as stationary phases in LC. The suspension method is less performed for MIP synthesis compared with bulk and precipitation methods and this may be a result of the need of a suspension medium that is either polar (water) or expensive (perfluorocarbon fluids).
Multi-Step Swelling Polymerization
To control the size distribution and the shape of the MIP particles more efficiently and to decrease the material loss in the form of fine particles, a polymerization method based on the swelling of uniformly sized particles was developed. The two-step swelling method was widely used by Haginaka's group for the synthesis of imprinted beads using different templates (57–60). It is a technique that enables the production of monodispersed spherical polymer particles over a size range 5–100 μm. It involves the production of uniformly sized polystyrene beads (called seeds or "shape template") of about 1 μm diameter by emulsifier-free emulsion polymerization.
These seeds are used for the two subsequent swelling steps. In the first step, called activation, the seeds are swollen using an aqueous micro-emulsion of a free radical initiator and a highly water-insoluble solvent (a low molecular weight activating solvent such as dibutyl phthalate or chlorodecane). This step is completed once all the emulsion droplets of the activating solvent are adsorbed into the seed particles. The swollen particles dispersion is added to a second aqueous dispersion of the polymerization mixture.
The mixture is again stirred until the droplets of polymerization mixture are adsorbed on the swollen particles. After the completion of the second swelling step, the polymerization reaction is initiated by keeping a gentle stirring. After 24 h, the polymer beads have to be washed with a range of organic solvents. The shape of the original seeds is preserved but their size increases considerably (5–100 times larger). The final beads size can be controlled by changing the levels of activating solvent and the volume ratios of the different dispersion phases. The chemical yield of beads prepared by two-step swelling polymerization is very high: greater than 88% (58).
Haginaka's group was the first to report aqueous two-step swelling applied to molecular imprinting of diaminophthalenes in 1994 (58). The imprinting effects were good, even with an aqueous dispersion phase, which inhibits hydrogen bonding between the template and the monomers. This is because the interactions developed between the template and the functional monomers are hydrophobic and based on strong ion-pair interactions. A bisphenol A (BPA) MIP obtained by multi-step swelling was directly integrated into the LC system as an on-line pretreatment column and connected to an electrochemical detector.
The direct determination of BPA in lake water samples at 10 parts per trillion (ppt) level was accomplished (59). Recently, Haginaka and colleagues reported a smart idea: they prepared uniform-sized MIP beads selectively modified with hydrophilic external layer, a restricted access media (10,15,28,61).
The obtained beads are called RAM–MIP, the RAM will exclude the macromolecules and the MIP will selectively retain the target analyte. A RAM–MIP imprinted with (S)-naproxen was used for the on-line direct determination of ibuprofen in serum samples (10).
Although the multi-step swelling seems to be the most suitable method for the production of uniform imprinted beads with a high yield, it is also one of the more laborious and complex procedures. The peak tailing and broadening are still observed using this polymerization method (57,58). This is because the heterogeneity of binding sites do not seem to be improved (62).
In-Situ Prepared Polymer Monoliths
Over the last decade, the constant analytical needs for lower detection limits (DLs), higher sensitivity and higher resolution has revolutionized chemistry. Miniaturized analytical devices have matured considerably since the initial lab-on-a-chip concept was proposed (63). Several laboratory procedures, including sample preparation, analytical separation, detection and quantification are integrated onto a single microchip named micro-total analysis systems (μ-TAS). The potential benefits of miniaturization are enormous, following the principle that small-scale processes consume less time, samples and reagents (64).
The miniaturization had a tremendous impact on MIP technology. The miniaturized MIP format attracted great interest because it may realize the use of templates or monomers for the molecular imprinting, which are expensive or otherwise difficult to achieve.
The advantages of the miniaturized MIP are numerous and include the ability to analyze minute samples, speed of analysis and reduced quantities of hazardous material and associated waste compared with the conventional MIP format where particularly high concentrations of the template are required. The introduction of molecularly imprinted-based phases in miniaturized systems can be achieved by packing a capillary column with MIP particles (65), but it requires frits, which are difficult to fabricate (66). The second approach consists of the in-situ polymerization procedure that leads to the synthesis of monolith (67-71).
Monolithic columns are continuous porous rods of stationary phase (72), MIP monoliths were mainly developed for capillary electrochromatography (CEC). The intrinsic higher efficiency and the enhanced flow dynamics of the CEC, combined with the higher selectivity of the MIP, are very attractive for the separation of chiral compounds (69,71). MIP monoliths are covalently anchored to the inner wall of fused-silica capillaries so that the use of retaining frits is unnecessary. Briefly, a typical procedure starts by the activation of the inner surface of the silica capillary using 3-(trimethoxysilyl)propyl methacrylate for a better attachment of the monolith (73). The capillary is then filled with the polymerization mixture, then the polymerization is performed by placing the capillary under a UV source or in heating bath (high temperature). When the polymerization is achieved, the capillary is flushed with a range of solvents to remove remaining reagents and to equilibrate it for later use.
This polymerization method is simple and quick compared with the procedures described previously. It enables the preparation of MIP monolith within 3 h. The success of the synthesis relies on the presence of macropores to provide good flow-through properties and on the simultaneous generation of the selective binding sites. Schweitz and colleagues (69) showed that the MIP monolith could be rendered porous by the use of iso-octane as co-porogen. Different parameters are controlling both the distribution of pores and the recognition properties, thus a careful optimization based on chemometric concepts seems interesting. Currently research is focused on the use of MIP monoliths as SPE sorbents. It has to be mentioned that there is a great attention to test and to optimize the MIP monolith format (67).
The future trends in the field of molecular imprinting goes toward the introduction of MIP into miniaturized devices (74,75). A recent report on this topic demonstrates the success of the combination of microfluidics, MIP and electrochemical sensing techniques for the selective detection of morphine (75).
Conclusion
This article demonstrated that molecular imprinting technology can solve some of the current analytical challenges for a SPE sorbent for the treatment of complex matrices, particularly when MIPs are used. Most MIPs previously produced for SPE were prepared in bulk. New polymerization strategies such as precipitation, suspension polymerization and two-step swelling were described.
These approaches are beneficial because they improve the shape of the MIP particles, the size range distribution, the reaction yield and are less time consuming. Current research is focusing on the improvement of the accessibility to binding sites to develop sorbents with higher affinity that can be routinely used in aqueous media.
There is also a need for research into molecular imprinting technology for the extraction of polar compounds that are mainly soluble in aqueous media. There is also an increasing demand for the development of MIP for macromolecules such as proteins. MIPs are most often produced at the gram level but research is turning towards their production at the process level to answer industrials needs. They have been produced at the kilogram level so their synthesis at the multi-kilogram level seems possible. Moreover, the outstanding stability of MIPs, their lower cost of development and their higher capacity give them a number of advantages over immunosorbents.
References
(1) N. Delaunay-Bertonicini, V. Pichon and M.-C Hennion, LCGC Eur. 14(3) 162–172 (2001).
(2) M.-C. Hennion and V. Pichon, J. Chromatogr. A 1000(1–2), 29–52 (2003).
(3) F. Chapuis,V. Pichon and M.C. Hennion, LCGC Eur. 17(7), 408–417 (2004).
(4) B. Sellergren, Molecularly imprinted polymers, man-made mimics of antibodies and their applications in analytical chemistry, (Elsevier, Amsterdam, The Netherlands, 2001) Vol. 23.
(5) B. Sellergren, Anal. Chem . 66(9) 1578–1582 (1994).
(6) V. Pichon, J Chromatogr. A , 1152 (1–2). 41–53 (2007).
(7) V. Pichon and K. Haupt, J. Liq. Chromatogr. Rel. Technol. 29, 989–1023 (2006).
(8) E. Caro et al., J. Chromatogr. A 995 (1–2), 233–238 (2003).
(9) S.Y Feng et al., J. Chromatogr. A 1027 (1–2), 155–160 (2004).
(10) J. Haginaka and H. Sanbe, Anal. Chem . 72 (21), 5206–5210 (2000).
(11) R. Koeber et al., Anal. Chem . 73(11), 2437–2444 (2001).
(12) E.P.C. Lai and S.G. Wu, Anal. Chim. Acta 481(2), 165–174 (2003).
(13) N. Masque et al., Anal. Chem. 72(17), 4122–4126 (2000).
(14) M.L. Mena et al., Anal. Chim. Acta 451(2), 297–304 (2002).
(15) H. Sanbe and J. Haginaka, The Analyst 128, 593–597 (2003).
(16) F.G. Tamayo, J. L. Casillas and A. Martin-Esteban, Anal. Chim. Acta 482(2), 165–173 (2003).
(17) G. Theodoridis et al., J. Chromatogr. 1030 (1–2), 69–76 (2004).
(18) S.G. Wu, E.P.C. Lai and P.M. Mayer, J. Pharm. Biomed. Anal. 36(3), 483–490 (2004).
(19) J. Xie, L. Zhu and X. Xu, Anal. Chem . 74 (10), 2352–2360 (2002).
(20) M. Zhang et al., J. Chromatogr. A 984(2), 173–183 (2003).
(21) V. Pichon, J. Chromatogr. A , 885(1–2), 195–215 (2000).
(22) K. Yoshizako et al., Anal. Chem . 70(2), 386–389 (1998).
(23) F. Chapuis, J. Chromatogr. B 804(1), 93–101 (2004).
(24) L.I. Andersson et al., Anal. Chim. Acta 526 (2), 147–154 (2004).
(25) F. Chapuis et al., J. Chromatogr. A 1135(2), 127–134 (2006).
(26) M. Komiyama et al., Molecular imprinting, from fundamentals to applications (Wiley-VCH, Germany, 2002).
(27) C. Crescenzi et al., Anal. Chem . 73 (10), 2171–2177 (2001).
(28) H. Sambe et al., J. Chromatogr. A 1134(1–2), 16–23 (2006).
(29) E. Caro, J. Chromatogr. A 1047(2), 175–180 (2004).
(30) F. Chapuis et al., J. Chromatogr. A 999 (1–2), 23–33 (2003).
(31) J.P Lai, R. Niessner and D. Knopp, Anal. Chim. Acta 522(2), 137–144 (2004).
(32) O. Bruggemann et al., Anal. Chim. Acta , 504(1), 81–88 (2004).
(33) N. M Maier et al., J. Chromatogr. B , 804(1), 103–111 (2004).
(34) K. Farrington, E. Magner and F. Regan, Anal. Chim. Acta , 566(1), 60–68 (2006).
(35) F. Puoci, et al., Food Chem. 93(2), 349–353 (2005).
(36) C. Chassaing et al., J. Chromatogr. B 804(1) 71–81 (2004).
(37) P.D Martin et al., J. Pharm. Biomed. Anal. 35(5), 1231–1239 (2004)
(38) K. Moller et al., J. Chromatogr. B: Analytical Technologies in the Biomedical and Life Sciences , 811(2), 171–176 (2004).
(39) E. Caro et al., J. Chromatogr. B 813(1–2), 137–143 (2004).
(40) M.T. Muldoon and L.H. Stanker, Anal. Chem. 69(5), 803–808 (1997).
(41) G. Brambilla et al., J. B: Biomedical Sciences and Applications 759(1), 27–32 (2001).
(42) L.Q Lin et al., Anal. Chim. Acta , 561(1–2), 178–182 (2006).
(43) E. Caro et al., TrAC Trends in Analytical Chemistry 25(2),143–154 (2006).
(44) L.I. Andersson, J. Chromatogr. B 739(1), 163–173 (2000).
(45) O. Bruggemann et al., J. Chromatogr. A , 889(1–2), 15–24 (2000).
(46) F.G. Tamayo et al., J. Chromatogr. A , 1069(2), 173–181 (2005).
(47) C. Cacho et al., J. Chromatogr. B , 802(2), 347–353 (2004).
(48) N. Perez-Moral and A.G. Mayes, Anal. Chim. Acta , 504(1), 15–21 (2004).
(49) C. Cacho et al., J. Chromatogr. A 1114(2), 255–262 (2006).
(50) P.J. Dowding and B. Vincent, Colloids and Surfaces A: Physicochemical and Engineering Aspects, 161 (2), 259–269 (2000).
(51) A. G. Mayes and K. Mosbach, Anal. Chem . 68(21), 3769–3774 (1996).
(52) R. J Ansell and K. Mosbach, J. Chromatogr. A , 787(1–2), 55–66 (1997).
(53) F.Wolf, B. Hoffbauer and S. Eckert, Plaste Kautsh , 19, 26–31 (1972).
(54) A. Guyot, In Synthesis and separations using functional polymers; D.C. Sherington, P. Hodge, Eds (John Wiley and Sons: London, UK, 1988).
(55) M Munger and E. Tromsdorf, In Polymerization processes , C.E. Schildkecht, I. Skeist, Eds. (Wiley Interscience, New York, USA, 1977).
(56) N. Perez-Moral, A.G. Mayes, Biosensors and Bioelectronics 21(9), 1798–1803 (2006).
(57) J. Haginaka and C. Kagawa, J. Chromatogr. B 804(1), 19–24 (2004).
(58) K. Hosoya et al., J. Chromatogr. A 728(1–2),139–147 (1996).
(59) Y. Watabe et al., J. Chromatogr. 1032(1–2), 45–49 (2004).
(60) M. Nakamura et al., J. Pharm. Biomed. Anal. 37(2), 231–237 (2005).
(61) H. Sanbe et al., The Analyst 130, 38–40 (2005).
(62) B. Sellergren and K.J. Shea, J. Chromatogr. A 690(1), 29–39 (1995).
(63) A. Manz, N. Graber and H.M. Widmer, Sensors and Actuators B: Chemical, 1 (1–6), 244–248 (1990).
(64) D.R. Reyes et al., Anal. Chem . 74(12), 2623–2636 (2002).
(65) J.M. Lin, K. Uchiyama and T. Hobo, Chromatographia V47(11), 625–629 (1998).
(66) M. Quaglia et al., J. Chromatogr. A 1044(1–2), 53–66 (2004).
(67) J. Courtois et al., J. Chromatogr. A 1109(1), 92–99 (2006).
(68) C.C. Lin, G.R. Wang and C.Y. Liu, Anal. Chim. Acta 572(2), 197–204 (2006).
(69) L. Schweitz, L.I. Andersson and S. Nilsson, J. Chromatogr. A 792(1–2), 401–409 (1997).
(70) L. Schweitz, L.I. Andersson and S. Nilsson, Anal. Chem. 69(6), 1179–1183 (1997).
(71) L. Schweitz, L.I. Andersson and S. Nilsson, Anal. Chim. Acta 435(1), 43–47 (2001).
(72) F. Svec, T.B. Tennikova and Z. Deyl, Monolithic Materials: Preparation, Properties and Applications (Elsevier, Amsterdam, The Netherlands, 2003).
(73) S. Hjerten, J. Chromatogr. A 347, 191–198 (1985).
(74) D. He et al., Talanta 69(5), 1215–1220 (2006).
(75) C. H. Weng et al., Sensors and Actuators B, 121(2), 576–582 (2007).
(76) T. Pap et al., J. Chromatogr. A 973(1–2), 1–12 (2002).
Faten BelHadj-Kaabi is a third year PhD student at the Department of Environmental and Analytical Chemistry (UMR CNRS 7121) at the Ecole Supérieure de Physique et Chimie Industrielles, Paris, France, under the supervision of Dr Valerie Pichon.
Valerie Pichon is assistant professor in the Department of Environmental and Analytical Chemistry (UMR CNRS 7121) at the Ecole Supérieure de Physique et Chimie Industrielles, Paris, France.

















Thermodynamic Insights into Organic Solvent Extraction for Chemical Analysis of Medical Devices
April 16th 2025A new study, published by a researcher from Chemical Characterization Solutions in Minnesota, explored a new approach for sample preparation for the chemical characterization of medical devices.

Sorbonne Researchers Develop Miniaturized GC Detector for VOC Analysis
April 16th 2025A team of scientists from the Paris university developed and optimized MAVERIC, a miniaturized and autonomous gas chromatography (GC) system coupled to a nano-gravimetric detector (NGD) based on a NEMS (nano-electromechanical-system) resonator.

