Middle-Up Characterization of the Monoclonal Antibody Infliximab by Capillary Zone Electrophoresis–Mass Spectrometry
LCGC Europe


CZE–ESI‑TOF‑MS for the characterization of the mAb infliximab and its variants is presented. Infliximab was analyzed using a middle-up approach involving either reduction or digestion with the enzyme IdeS. A multilayer capillary coating of PB-DS‑PB in combination with a background electrolyte of 40% acetic acid provided efficient separation of the obtained antibody fragments, that is, LC and HC, as well as F(ab’)2 and Fc/2 parts. C-terminal lysine variants were also resolved. Recorded mass spectra of HC and Fc/2 fragments permitted assignment of 13 glycoforms and provided a quantitative profile, with G0F the most abundant glycoform (~50%). CZE–ESI-TOF-MS represents an efficient means for the straightforward analysis of a monoclonal antibody and its proteoforms.
Capillary zone electrophoresis-electrospray ionization time-of-flight mass spectrometry (CZE–ESIâTOFâMS) for the characterization of the monoclonal antibody (mAb) infliximab and its variants is presented. Infliximab was analyzed using a middle-up approach involving either reduction or digestion with the enzyme IdeS. A multilayer capillary coating of Polybrene-dextran sulfate-Polybrene (PB-DSâPB) in combination with a background electrolyte of 40% acetic acid provided efficient separation of the obtained antibody fragments, that is, light chain (LC) and heavy chain (HC), as well as F(ab’)2 and Fc/2 parts. C-terminal lysine variants were also resolved. Recorded mass spectra of HC and Fc/2 fragments permitted assignment of 13 glycoforms and provided a quantitative profile, with G0F the most abundant glycoform (~50%). CZE–ESI-TOF-MS represents an efficient means for the straightforward analysis of a monoclonal antibody and its proteoforms.
Monoclonal antibodies (mAbs) have taken a prominent and increasingly important position in today’s drug development and production, providing high specificity and limited side effects in the treatment of lifeâthreatening diseases. The production of a mAb using mammalian cells will often result in a heterogeneous product, as it will undergo postâtranslational modifications (PTMs) (1).
Moreover, potential degradation or rearrangements at different stages of the production, purification, and storage may lead to structural variants (1). N-glycosylation is an important source of heterogeneity of mAbs and most commonly occurs in the crystallizable (Fc) domain of the heavy chain (HC) (2). Fc glycosylation is fundamental to provide the mAb with the proper effector functions and affects quality, pharmacokinetics, immunogenicity, and potency of the drug. Glycosylation is considered a critical quality attribute (CQA) of the mAb (2). Antibody variants, resulting from asparagine and glutamine deamidation, methionine oxidation, C-terminal lysine truncation, or N-terminal pyroglutamation, are other sources of product heterogeneity (3), and should be assessed at different stages of the production.
Mass spectrometry (MS) has become a primary tool for the characterization of mAbs (4), yielding molecular weights of the main protein and variants, identification of PTMs, and glycosylation patterns. MS can be used for analysis at the intact mAb level using a top-down approach, or for the determination of peptides obtained after enzymatic digestion of the mAb (that is, bottomâup). The choice will mainly depend on the analytical question, for example, relating to overall protein heterogeneity or to specific locations of protein modification. Intact mAb analysis might be hindered by poor ionization efficiencies and lack of MS resolution. The so-called middle-up approach provides a practical alternative, facilitating the characterization of mAb heterogeneity by analyzing mAb subunits of 25–50 kDa (5). Before analysis, mAbs are cleaved by reduction of the intra-chain disulfide bridges (yielding light chain [LC] and heavy chain [HC]) or by enzymatic cleavage using papain or IdeS (yielding Fab and Fc fragments). Middle-up analysis decreases the overall complexity of the probed molecules, resulting in a more straightforward interpretation of the obtained MS data and allowing more accurate assessment of glycosylation profiles. It has been applied to the MS-based assessment of the conjugation degree of antibody conjugates (6), and for the analysis of degradation products in mAbs (7), modifications in HCs or LCs (8), and HC glycosylation (9).
When analyzing real-life samples (which are frequently mixtures), obtaining good-quality mass spectra of proteins often requires separation prior to MS detection. Several separation techniques can be employed for the separation of mAbs and their subunits (10,11). Charge heterogeneity can be assessed by ion exchange chromatography (IEC) and capillary isoelectric focusing (CIEF), whereas size-exclusion chromatography (SEC) and capillary gel electrophoresis (CGE) can be used for determination of size variants. Unfortunately, the mobile phases employed to obtain the highest separation efficiency by these methods often show serious incompatibilities with MS detection. Therefore, reversedâphase liquid chromatography (LC) is still most commonly used in combination with MS for mAb characterization (12), although reversed-phase LC may lack the selectivity to distinguish certain protein modifications. Recently, hydrophilic interaction liquid chromatography (HILIC)–MS has been shown to be very useful for resolving glycoforms of biopharmaceuticals, including mAbs (13). Capillary zone electrophoresis (CZE), on the other hand, has shown good potential for the separation of closely related charge variants and isoforms of proteins (14). In addition, CZE has the intrinsic capacity to produce narrow peaks for proteins and-nowadays-can be coupled rather easily to mass spectrometric detection. Consequently, over the last few years, the use of CZE–MS in middle-up and intact mAb analysis has increased. It has, for example, been used to separate mAb subunits and estimate their molecular weights (15), to identify PTMs on the intact and subunit level (16,17), and to make in-depth glycosylation profiles (18).
The aim of this article is to highlight the potential of CZE–MS for the middle-up characterization of mAbs. Infliximab was selected as a model compound because it comprises extensive variation in glycosylation as well as HC C-terminal lysine variants. Prior to CZE–MS analysis, infliximab was either reduced to yield its HCs and LCs, or digested using the proteolytic enzyme IdeS, resulting in its F(ab)2 and Fc/2 subunits. CZE separation, CZE–MS interfacing, and MS conditions were carefully optimized to achieve detailed glycoform and C-terminal lysine profiles of the infliximab fragments.
Materials and Methods
Chemicals: Polybrene (hexadimethrine bromide, PB; average Mw = 15,000), dextran sulfate sodium salt (DS; average Mw > 500,000), methanol, propanâ2âol (IPA), ethylene glycol (99.5%), and DL-dithiothreitol (99.0%) were purchased from Sigma Aldrich. Acetic acid (99.8%) and ammonium hydroxide (25% v/v) were obtained from Merck. A formulation of infliximab (Remicade) comprised sucrose, sodium phosphate, and polysorbate 80. FragIT MicroSpin columns containing immunoglobulinâdegrading enzyme of Streptococcus pyogenes (IdeS; Genovis) were used for mAb digestion. Ultra-pure water was produced by a Milli-Q system (Millipore). Background electrolyte (BGE) was prepared daily by diluting acetic acid with water to a final concentration of 40% v/v (6.9 M, ~pH 2.0).
Sample Preparation: Reduction of protein disulfide bonds was accomplished by incubating the infliximab formulation with 50-mM dithiothreitol (DTT) for 10 min at 37 °C. The reaction was quenched with acetic acid (final concentration, 1% v/v). Digestion with IdeS was performed following the FragIT MicroSpin (Genovis) kit instructions. All final reaction mixtures were centrifuged three times against six volumes of water using Vivaspin 3 kDa MWCO filter tubes (GE Healthcare) and applying 15000 × g at 25 °C. The sample residue was reconstituted in water and yielded a final concentration of 5 mg/mL of infliximab.
CE System: The CZE experiments were performed on a PA 800plus capillary electrophoresis system (Beckman Coulter). Fused-silica capillaries had a total length of 80 cm and 50 μm internal diameter (i.d.) × 375 μm outside diameter (o.d.) (Polymicro Technologies). New capillaries were subsequently rinsed with methanol (5 min), 1 M NaOH (30 min), and water (10 min) at 20 psi. After conditioning, Polybrene-dextran sulfate-Polybrene (PB-DSâPB) triple layer coating was applied using the procedure described elsewhere (19). Samples were injected hydrodynamically at 1.5 psi for 5 s (~1% of the capillary volume). A voltage of -30 kV was applied, and the cartridge temperature was maintained at 25 °C. CZE–UV measurements were conducted in a 60 cm capillary (50 cm to the detector) treated in the same way as described earlier. The separation voltage was -25 kV and the detection wavelength was 214 nm.
Mass Spectrometry: A micrOTOF QII orthogonalâaccelerated quadrupole time-of-flight (QTOF) mass spectrometer (Bruker Daltonics) was used for detection of mAb fragments. CZE–MS coupling was achieved via a co-axial sheath-liquid interface (Agilent Technologies). Sheath liquid consisting of 1:1 IPA and water (v/v) was delivered by a 2.5 mL gasâtight syringe (Hamilton) at a flow rate of 180 µL/h using a syringe pump (KD Scientific Inc.). Electrospray ionization (ESI) in positive mode was achieved applying a voltage of -4.5 kV. Other settings were: nebulizer pressure, 0.4 bar; dry gas flow rate, 4.0 L/min; dry gas temperature, 190 °C; ion energy, 5 eV; collision energy, 10 eV; in-source collisionâinduced dissociation, 0 eV. CZE–MS data were analyzed using Bruker Daltonics Data Analysis software. Molecular weight determinations of proteins were performed using the “Charge Deconvolution” utility of the software.
Results and Discussion
The model compound infliximab is an immunoglobulin G1 (IgG1) therapeutic antibody comprised of two LCs (214 amino acids each) and two HCs (450 amino acids each) interconnected by four disulfide bridges. From the amino acid sequence of the LC and HC (Figure S1, see supplementary material: http://www.chromatographyonline.com/supplementary-information-middle-characterization-monoclonal-antibody-infliximab-capillary-zone-elec), the theoretical molecular weights and isoelectric points of the protein subunits were derived.


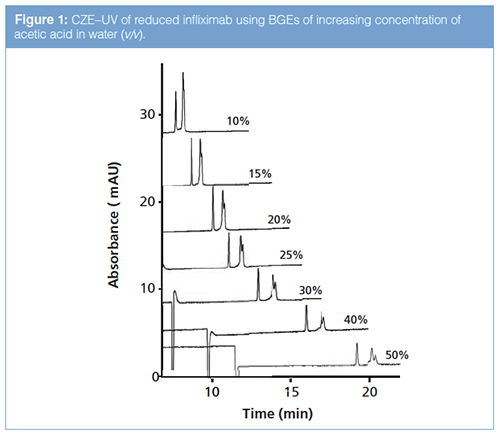
CZE–MS Conditions: Reduction of protein disulfide bonds using DTT represents a straightforward way to generate LC (~25 kDa) and HC (~50 kDa) fragments of a mAb. CZEâUV analysis of a reduced infliximab sample was used for optimization of the separation conditions. A BGE of 0.5% (v/v) acetic acid (pH 3.0) was used as the starting point, in which both the LC (pI 5.8) and HC (pI 8.2) are net positively charged. In order to prevent protein adsorption to the capillary inner wall, a positively charged PB-DS-PB coating was applied, yielding an anodic electroosmotic flow. This multilayer coating is fully MS-compatible and can allow highly efficient protein CZE (19). Using 0.5% acetic acid as BGE, a partial separation of the mAb fragments was obtained; this was improved by increasing the concentration of acetic acid. Analysis of the reduced infliximab employing a BGE of 10% (v/v) acetic acid resulted in two well-resolved, narrow, and symmetric peaks (Figure 1). Further increase of the acetic acid concentration provided an enhanced protein separation with the later migrating peak resolving into two bands. A BGE of 40% (v/v) acetic acid (pH 2.0) was selected for further CZE–MS experiments. The addition of methanol, IPA, or ethylene glycol up to 10% (v/v) led to longer protein migration times, but did not improve the separation any further.
CZE–MS coupling was accomplished using a coaxial sheath liquid sprayer and the reduced infliximab sample was analyzed. Sheath liquids consisting of either methanol or IPA in a 1:1 ratio (v/v) with water containing 0.1% acetic acid (v/v) were considered. With both sheath liquids, reduced infliximab yielded three main peaks, such as those observed in CE–UV. From the deconvoluted mass spectra it could be concluded that the first and second migrating peaks were the LC and HC, respectively. The mass obtained for the third peak corresponded with the HC with a C-terminal lysine (HCK). The extra lysine added one positive charge to the HC, resulting in an increase of the protein migration time in the applied CZE system. More detailed interpretation of the obtained mass spectra is given in the section “Middle-Up Characterization”.
Sheath liquids containing IPA yielded more than twofold larger protein peak areas when compared to sheath liquids with methanol. Further evaluation of the sheath liquid composition indicated that acetic acid (0–1% v/v tested) decreased the protein signals up to 25% and that the IPA percentage (25–75% v/v tested) was optimal at 50%. The flow rate of the optimum sheath liquid of 1:1 IPA–water (v/v) was tested in the range 1–5 µL/min. Between 2–3.5 µL/min, stable baselines and no change in LC and HC peak areas were observed. Above 3.5 µL/min, protein signals decreased because of effluent dilution, whereas below 2 µL/min unstable sprays were obtained.
The PB-DS-PB coating in combination with a BGE of 40% (v/v) acetic acid and a sheath liquid of 1:1 IPA–water (v/v) at the flow rate 3 µL/min provided stable separation and favourable detection conditions. The repeatability of the method was examined by successive CZE–MS analyses (n = 5) of reduced infliximab. Migration times and peak areas of the LC, HC, and HCK subunits were derived from the extracted-ion electropherograms (EIEs) constructed for the most intense ion observed in the mass spectra of the respective species. Relative standard deviations (RSDs) for migration times were below 2.0%, whereas peak area RSDs did not exceed 7.5%. Plate numbers obtained for the LC and HC peaks were 220,000 and 90,000, respectively.
Middle-Up Characterization:Reduced Infliximab: CZE–MS analysis of reduced infliximab yielded three main peaks (Figure 2[a]) of which the mass spectra are depicted in Figures S2(a–c) (see supplementary material: http://www.chromatographyonline.com/supplementary-information-middle-characterization-monoclonal-antibody-infliximab-capillary-zone-elec). The deconvoluted mass spectrum of the first peak (Figure 2[b]) reveals a main molecular mass of 23433.8 Da, which is in good agreement with the theoretical mass of the infliximab LC when taking interâchain disulfide bond reduction into account.

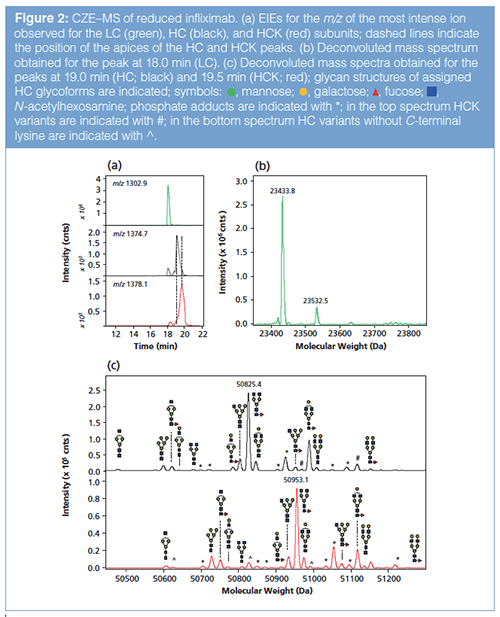
Considering the observed mass, the intra-chain bonds are still intact, most probably because no denaturing agent was used prior to DTT addition. The minor signal at 23532.5 Da (Figure 2[b]) can be ascribed to a phosphate adduct of the LC (20). For most proteins analyzed with CZE–MS, minor phosphate adducts were observed, which might be related to an ionâsource contamination.
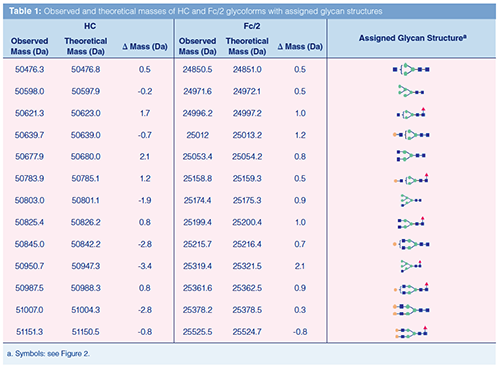
EIEs derived for the second and third peak show a partial separation of the two involved species (Figure 2[a]). The deconvoluted mass spectrum of the second peak (Figure 2[c]) shows a main signal at 50825.4 Da, which corresponds to the G0F glycoform of the intact HC (that is, no intraâchain disulfide reduction) (Table 1). From the other masses observed in the deconvoluted spectrum, 12 additional glycoforms could be assigned based on known glycan masses (Figure 2[c] top, Table 1). The glycoforms varied between biantenary complex glycans and high-mannoseâtype glycans with and without core fucosylation. Further minor signals originated from phosphate adducts or HCK variants that were not fully separated from the HC species. For the third peak, a main mass of 50953.1 Da was obtained (Figure 2[c] bottom), which matches the HC G0F glycoform including a C-terminal lysine. Indeed, for the other 12 HC glycoforms, C-terminal lysine variants were observed in the deconvoluted mass spectrum (Figure 2[c] bottom; Table S1, see supplementary material: http://www.chromatographyonline.com/supplementary-information-middle-characterization-monoclonal-antibody-infliximab-capillary-zone-elec).

IdeS-Digested Infliximab: CZE–MS analysis of IdeS-digested infliximab using the same conditions as stated above yielded three baseline-separated peaks (Figure 3[a]) with corresponding mass spectra (Figure S2[d–f], see supplementary material: http://www.chromatographyonline.com/supplementary-information-middle-characterization-monoclonal-antibody-infliximab-capillary-zone-elec). The deconvoluted mass spectrum of the first peak showed a main molecular mass of 98152.5 Da (Figure 3[b]), which agrees with the theoretical mass of the F(ab’)2 fragment with intact inter-chain disulfide bonds. Some minor signals were observed, from which one (98250 Da) could be assigned to a phosphate adduct of the F(ab’)2 subunit.

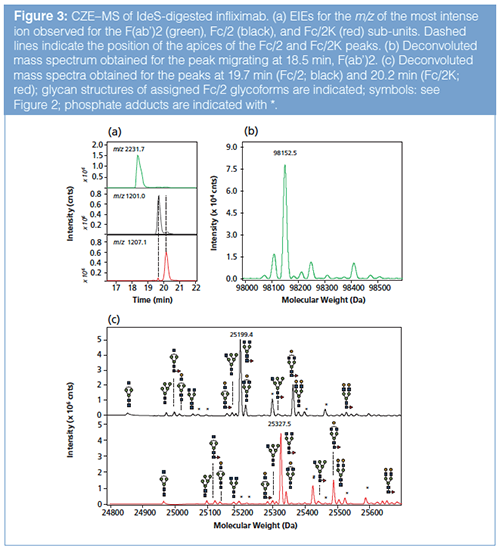
Deconvolution of the mass spectra recorded for the second and third peak yielded main molecular masses of 25199.4 and 25327.5 Da, respectively (Figure 3[c]). The second peak thus comprises the Fc/2 fragment of infliximab bearing the G0F glycan structure. The main mass of the third peak corresponds to the Fc/2 G0F glycoform with a C-terminal lysine (Fc/2K). Based on the observed masses, in addition to the main glycoform, 12 other glycoforms could be assigned for both the native Fc/2 fragment (Table 1) and the Fc/2 lysine variants (Table S1, see supplementary material: http://www.chromatographyonline.com/supplementary-information-middle-characterization-monoclonal-antibody-infliximab-capillary-zone-elec). The assigned glycoforms nicely matched those observed for reduced infliximab. The Fc/2 and Fc/2K subunits were baseline separated, so no Fc/2K signals were observed in the Fc/2 spectrum and vice versa.
Considerations and Comparisons: Good mass resolution is required to resolve comigrating compounds. TOF-MS provided a mass resolution of 12,000, allowing differentiation of mass differences of about 2.1 Da and 4.2 Da among proteins of 25 and 50 kDa, respectively. This resolving power is quite sufficient for distinguishing different glycoforms of HC or Fc subunits. However, minor mass modifications, such as the reduction of one disulfide bridge (Δ = 2 Da) or deamidation (Δ = 1 Da), cannot be resolved. These result in broadening of the MS signal and may contribute to the uncertainty of the measured mass. In the present study, observed mass deviations from theoretical values did not exceed 2.1 and 3.4 Da for the Fc/2 and HC fragments, respectively. This means that the observed molecular masses can be reliably assigned to the indicated glycoforms. Indeed, the assigned structures are in accordance with glycans typically found for IgG1 antibodies. The majority of the glycoforms were previously indicated for infliximab applying either bottom-up (21), released glycan (22), or middle-up (23) approaches.
From the deconvoluted mass spectra of the HC and Fc/2, relative intensities for the assigned glycoforms can be derived assuming an equimolar response per glycoform (Figure 4). The main glycoform (G0F) represents more than 50% of the total signal, whereas the low abundant species are around 0.5%. This illustrates the excellent dynamic range of the developed method. When comparing the glycosylation profiles obtained for the HC and Fc/2 subunits, only minor differences were observed in relative intensities for the glycoforms, confirming that the responses reflect glycoform composition.

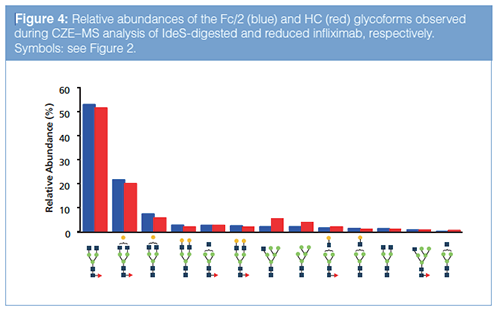
In comparison with the middleâup approach employing reduction, IdeS digestion exhibited increased MS signal intensity and better CZE separation of the subunits. The smaller size of the Fc/2 fragments relative to the HC fragments yield more efficient ionization and narrower charge state distributions, providing higher sensitivity and better resolved mass spectra. From the CZE point of view, differences caused by modifications are relatively larger for Fc/2 fragments and therefore induce more efficient separations. The Fc/2 and Fc/2K fragments were baseline separated by CZE, whereas this was not the case for the HC and HCK fragments. Moreover, no glycoform separation was obtained for the HC fragments, whereas partial separation of Fc/2 glycoforms was observed (Figure S3, see supplementary material: http://www.chromatographyonline.com/supplementary-information-middle-characterization-monoclonal-antibody-infliximab-capillary-zone-elec). As the glycans are neutral, the differences in electrophoretic mobility of the Fc/2 glycoforms can be ascribed to size differences. Similar observations were made in previous CZE studies of intact glycoproteins (18,24).
Conclusions
CZE–MS employing a positively charged, noncovalent coated capillary and a sheath-liquid interface has been evaluated for the middle-up analysis of the antibody infliximab. The developed method allowed for fast screening of antibody heterogeneity, exploiting the combined resolution of CZE and TOF-MS for reliable assignment of C-terminal lysine variants and glycoforms. Independent of the middle-up approach chosen (that is, reduction or IdeS digestion), separation of the subunits was obtained under the same conditions. Moreover, highly similar glycoprofiles were obtained in both cases. Overall, this work clearly highlights the potential of the developed CZE–MS method for the assessment of mAb microheterogeneity.
Acknowledgement
Financial support from the European Social Fund and the State Budget of the Czech Republic, Project no. CZ.1.07/2.3.00/30.0061, is gratefully acknowledged.
References
- X. Wang, Z. An, W. Luo, N. Xia, and Q. Zhao, Protein Cell9, 74–85 (2018).
- F. Cymer, H. Beck, A. Rohde, and D. Reusch, Biologicals52, 1–11 (2018).
- S. Fekete, A.L. Gassner, S. Rudaz, J. Schappler, and D. Guillarme, Trends in Analytical Chemistry42, 74–83 (2013).
- H. Zhang, W. Cui, and M.L. Gross, FEBS Letters588, 308–317 (2014).
- A. Beck, F. Debaene, H. Diemer, E. Wagner-Rousset, O. Colas, A. van Dorsselaer, and S. Cianférani, Journal of Mass Spectrometry50, 285–297 (2015).
- E. Wagner-Rousset, M.C. Janin-Bussat, O. Colas, M. Excoffier, D. Ayoub, J.F. Haeuw, I. Rilatt, M. Perez, N. Corvaia, and A. Beck, Mabs6, 173–184 (2014).
- H. Lau, D. Pace, B.X. Yan, T. McGrath, S. Smallwood, K. Patel, J. Park, S.S. Park, and R.F. Latypov, Journal of Chromatography B878, 868–876 (2010).
- J. Zhang, H.B. Liu, and V. Katta, Journal of Mass Spectrometry45, 112–120 (2010).
- E. Wagner-Rousset, A. Bednarczyk, M.C. Bussat, O. Colas, N. Corvaia, C. Schaeffer, A. Van Dorsselaer, and A. Beck, Journal of Chromatography B872, 23–37 (2008).
- A.L. Capriotti, C. Cavaliere, P. Foglia, R. Samperi, and A. Laganà, Journal of Chromatography A1218, 8760–8776 (2011).
- M. Tassi, J. De Vos, S. Chatterjee, F. Sobott, J. Bones, and S. Eeltink, Journal of Separation Science41, 125–144 (2018).
- B. Bobály, V. D’Atri, M. Lauber, A. Beck, D. Guillarme, and S. Fekete, Journal of Chromatography B1096, 1–10 (2018).
- E. Domínguez-Vega, S. Tengattini, C. Peintner, J. van Angeren, C. Temporini, R. Haselberg, G. Massolini, and G.W. Somsen, Talanta184, 375–381 (2018).
- R. Haselberg, G.J. de Jong, and G.W. Somsen, Journal of Chromatography A1159, 81–109 (2007).
- M. Han, B.M. Rock, J.T. Pearson, and D.A. Rock, Journal of Chromatography B1011, 24–32 (2016).
- A.M. Belov, L. Zang, R. Sebastiano, M.R. Santos, D.R. Bush, B.L. Karger, and A.R. Ivanov, Electrophoresis39, 2069–2082 (2018).
- K. Jooß, J. Hühner, S. Kiessig, B. Moritz, and C. Neusüß, Analytical and Bioanalytical Chemistry409, 6057–6067 (2017).
- R. Haselberg, Th. De Vijlder, R. Heukers, M.J. Smit, E.P. Romijn, G.W. Somsen, and E. Dominguez-Vega, Analytica Chimica Acta1044, 181–190 (2018).
- R. Haselberg, G.J. de Jong, and G.W. Somsen, Analytica Chimica Acta678, 128–134 (2010).
- S.K. Chowdhury, V. Katta, R. Beavis, and B.T. Chait, Journal of the American Society for Mass Spectrometry1, 382–388 (1990).
- J. Stadlmann, M. Pabst, D. Kolarich, R. Kunert, and F. Altmann, Proteomics 8, 2858–2871 (2008).
- C. Lee, M. Jeong, J.J. Lee, S. Seo, S.C. Cho, W. Zhang, and O. Jaquez, mAbs9, 968–977 (2017).
- M. Tsuda, Y. Otani, A. Yonezawa, S. Masui, Y. Ikemi, M. Denda, Y. Sato, S. Nakagawa, T. Omura, S. Imai, T. Nakagawa, M. Hayakari, and K. Matsubara, Biological and Pharmaceutical Bulletin41, 1716–1721 (2018).
- R. Haselberg, G.J. de Jong, and G.W. Somsen, Analytical Chemistry85, 2289–2296 (2013).
Klára Michal’ková works at the Laboratory of Environmental Biotechnology of the Academy of Sciences, Czech Republic, focusing on emerging organic pollutants decomposition in the environment using microorganisms. Prior to this, she gained extensive knowledge in the field of capillary electrophoresis and its hyphenation with mass spectrometry while obtaining her Ph.D. at the Faculty of Pharmacy of Charles University, Czech Republic.
Elena Dominguez Vega received her Ph.D. degree at the University of Alcala, Spain. Afterwards, she joined the group of Professor G.W. Somsen (Vrije Universiteit Amsterdam) where she worked on the development of new electrophoretic technologies to study protein heterogeneity, conformation, and affinity. Subsequently, she joined the Center for Proteomic and Metabolomics (Leiden University Medical Center) as senior researcher with a focus on MS-based methodologies to characterize intact proteins of pharmaceutical and biomedical interest.
Govert W. Somsen is a full professor of biomolecular analysis and analytical chemistry at the Vrije Universiteit Amsterdam, Netherlands. His current research interests include the compositional and conformational characterization of intact biomacromolecules, and the bioactivity screening of compounds in complex samples. His group have built up an internationally recognized expertise in the hyphenation of separation techniques with mass spectrometry and optical spectroscopy for the analysis of intact proteins.
Rob Haselberg is an assistant professor at the Vrije Universiteit Amsterdam where he focuses on the characterization of intact (bio)macromolecules, such as biopharmaceuticals and industrial polymers. For this, he develops and applies novel analytical approaches based on liquid chromatography, capillary electrophoresis, mass spectrometry, and optical detection.
Koen Sandra is the editor of “Biopharmaceutical Perspectives”. He is the Scientific Director at the Research Institute for Chromatography (RIC, Kortrijk, Belgium) and R&D Director at anaRIC biologics (Ghent, Belgium). He is also a member of LCGC Europe’s editorial advisory board. Direct correspondence about this column to the editor-in-chief, Alasdair Matheson, at alasdair.matheson@ubm.com
















Biopharmaceutical Characterization in the Age of Artificial Intelligence
May 13th 2025AI-powered tools are enhancing precision, efficiency, and decision-making in biopharmaceutical development. Recently, Jared Auclair and Anurag Rathore explored AI's evolving role in biopharmaceuticals in detail.


, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.



