Injecting Water onto a GC Column: Solving the Mystery of Poor Chromatography
Ethylene glycol is a particularly difficult compound to analyze because it is not easily extracted from water. Many environmental samples originate from water runoff at airports, where ethylene glycol is used as a de‑icing agent for airplanes during winter months. Hydraulic fracturing is a technique where pressurized fluid and sand or other solids (proppant) are used in gas drilling to allow gas extraction. Glycols are a common ingredient in most hydraulic fracturing fluid and play a key role in preventing emulsifications and stabilizing the solutions. The direct aqueous injection of ethylene glycol is challenging because it can be difficult to attain reproducibility and good peak shape. The large expansion volume of water can cause backflash, carryover can cause inconsistent results, and excess water can extinguish the flame ionization detection (FID) flame. This article describes a robust approach to analyze glycols in aqueous samples, which reduces downtime and maintains sensitivity.


Photo Credit: netsign33/Shutterstock.com

Ethylene glycol is a particularly difficult compound to analyze because it is not easily extracted from water. Many environmental samples originate from water runoff at airports, where ethylene glycol is used as a deâicing agent for airplanes during winter months. Hydraulic fracturing is a technique where pressurized fluid and sand or other solids (proppant) are used in gas drilling to allow gas extraction. Glycols are a common ingredient in most hydraulic fracturing fluid and play a key role in preventing emulsifications and stabilizing the solutions. The direct aqueous injection of ethylene glycol is challenging because it can be difficult to attain reproducibility and good peak shape. The large expansion volume of water can cause backflash, carryover can cause inconsistent results, and excess water can extinguish the flame ionization detection (FID) flame. This article describes a robust approach to analyze glycols in aqueous samples, which reduces downtime and maintains sensitivity. - Chris English, Restek, State College, Pennsylvania, USA
Many analysts have struggled with aqueous injections; especially to meet the required detection limits. Polar analytes are particularly challenging because alternative methods of analysis require derivatization or complex sample extraction procedures that may introduce a new set of challenges. One of the most notorious pairs of compounds to analyze are propylene (PG) and ethylene glycol (EG). Exposure to glycols can occur through environmental contamination of water with automobile antifreeze, aircraft de-icing liquids, and hydraulic fracturing fluid. These compounds have a significant difference: just 2–4 ounces of ethylene glycol can be fatal if ingested, whereas propylene glycol is commonly used in foods. The toxicity of ethylene glycol and potential for exposure from a wide range of sources makes reliable low-level quantification critical.
When I worked in an environmental laboratory, I struggled with tailing peaks, retention time shifts, and nonlinear calibration curves. Peak tailing was so severe that manual integration was necessary. After repeated injections, propylene and ethylene glycol would merge together making identification impossible. Maintenance was sometimes performed after only ten samples and required changing the liner and cutting and baking the column. The method called for a 1-µL splitless injection of standards and samples in water, which were introduced into a 4.0âmm internal diameter (i.d.) single gooseneck liner without wool. This approach assumes most of the sample will be transferred onto a 30 m × 0.53 mm, 1.0-μm polyethylene glycol (PEG) stationary phase. Using a large internal diameter liner allows for the rapid expansion of water vapour and is meant to reduce flashback; however, under our conditions water expands to 1867 µL in a liner with an effective internal volume of 493 µL. This means that not only are we losing a significant amount of sample, but water is also backflashing into the inlet lines and leaving sample residue throughout the injection port. This contamination can, over time, bleed out resulting in ghost peaks, carryover, and an elevated baseline.
Transferring all of the sample onto the column requires slow flows through the liner while active analytes are exposed to the high temperature of the injection port and compounds have longer contact time with active surfaces. Even stable compounds have wider peak widths compared to split analysis using this slow transfer. In the environmental laboratory, a dual column was used with one injection port and a guard column attached to a press-tight “Y” connector that split to a PEG column and a confirmation column, a trifluoropropyl phase.
This second phase is considered nonpolar relative to a PEG column and did not perform as well with water. Water does not properly focus on this phase and forms droplets at the head of the column, which can result in split peaks, shifting retention times, and in extreme examples may extinguish the flame ionization detector (FID). When everything was running smoothly our reporting limit was 10 ng on-column and subsequent work using a direct injection (uniliner) technique cut the reporting limit in half.
We revisited this analysis with the goal of reproducing some of the chromatographic struggles of my past and developing a new approach that will extend column lifetime, reduce maintenance, and achieve lower detection limits. Returning to splitless injection, we followed the same conditions as above. Initial chromatography using a PEG phase resulted in acceptable peak shape initially, but following 80 water injections in splitless mode produced those familiar broad peaks that over time move together into an indiscernible hump (Figure 1).

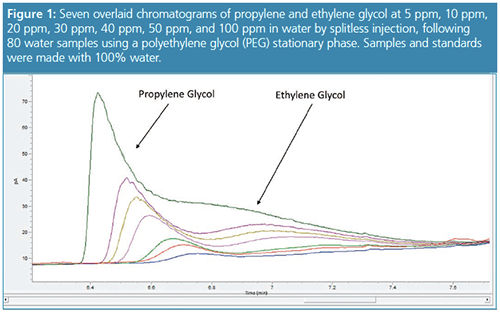
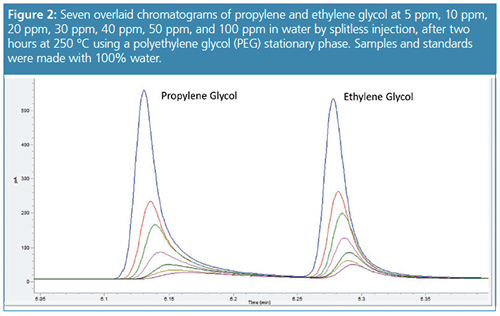
Our theory is that the hydrophilic nature of the PEG column and hydrogen bonding of the phase with water allows the column to retain moisture even at temperatures of 250 °C. The oven conditions used a final oven hold time of 250 °C for 2 min. After conditioning the column for two hours at 250 °C the peak shapes and retention times were restored to original performance (Figure 2). We were able to run 100% water samples and degrade the chromatography again over the course of 20 injections. An additional problem with 100% aqueous samples is the build-up of residue in the barrel of the syringe causing the syringe plunger to either become bent during injection or stuck causing an autosampler error (Figure 3). Another interesting trick other laboratories have used to maintain performance is to add methanol because it can assist in removing water from the column and can prevent autosampler syringe problems. In our next experiment we used 10:90 methanol–water to standards and water samples without baking out the column and maintained the gas chromatography (GC) program with a 2-min hold at 250 °C. Again, the column performance was restored with performance similar to Figure 2.



My colleague, Corby Hilliard, set out to develop a comprehensive in-house method development study by evaluating a variety of conditions that would result in reliable symmetrical peaks and stable retention times (1). A lifetime study was conducted on three PEG columns to determine if the method would hold up to repeated water injections and maintain chromatographic performance. The experimental design consisted of ten injections of 100% water (1 µL splitless) followed by an injection of a 50 µg/mL glycol standard (50:1 split injection, 1 ng onâcolumn). The injections were made in splitless mode to increase column exposure to water, creating a more severe test; the standard injection was made in split mode.
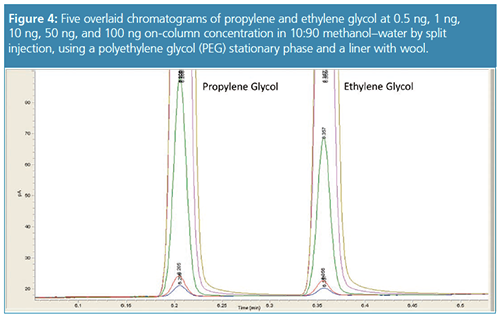
Peak symmetry was evaluated after every ten water injections and the procedure was repeated until the three columns had reached 600 water injections. The most notable departure from our previous work was the use of a high split ratio. It may seem counter-intuitive to vent 98% of the sample and expect better sensitivity, however, the study showed that a 1 µL 50:1 split injection into a liner containing wool eliminated injection port effects, decreased backflash, increased reproducibility, and produced better sensitivity. Using a high split ratio also allows the use of more nonpolar stationary phases. A trifluoropropyl phase column can be used as a confirmation column in split mode because the amount of water on-column is 1/50th of a µL. Since flows through the liner are 50 times higher than a splitless injection, the use of wool in a 4.0-mm-i.d. liner was critical for good reproducibility because the wool allows the sample to vaporize rapidly and completely. The standards were analyzed using split injection across a calibration range that went from 0.5-ng on-column to 100-ng on-column (Figure 4). While carryover is closely associated with splitless injections and the injection port in general, it can also be caused by sample residue in the syringe being transferred from one injection to another. If the syringe is not properly cleaned between analyses or if 100% water is used as the rinse solvent carryover will cause inconsistent results and may permanently damage the syringe plunger (Figure 3). Rinsing the syringe with 50:50 methanol–water three to six times between each injection will eliminate most sample residue and minimize the possibility of carryover. Preparing standards and samples in 10% methanol is recommended for routine analysis because it helps prevent the syringe needle from seizing up and more importantly reduces build-up of water on the PEG phase.

Conclusions
Analyzing highly polar analytes in water samples using split injection provides several advantages over the typical splitless approach: it reduces the risk of backflash, allows for better sensitivity, and also prevents problems with peak tailing and shifting retention times. By using this split injection method with a PEG column, very consistent chromatographic performance was obtained, even following the injection of 600 water samples. Peak symmetry measured after every 100 water samples averaged 0.95 for propylene glycol and 0.99 for ethylene glycol with percent relative standard deviations of 1.2% and 0.7%, respectively. A calibration curve with 0.5 ng, 1.0 ng, 10 ng, 50 ng, and 100 ng on-column had correlation coefficients of 0.9999 for both glycols. Split injection on PEG delivers fast, consistent sample transfer with less water on-column. The method produces symmetrical peaks that elute at stable retention times, which improves reproducibility and sensitivity for low-level aqueous samples.
Reference
- C. Hilliard and C. English, Reliable LowâLevel Analysis of Glycols in Water Using Split Injection, Application Note EVAN2873-UNV (2018).
Chris English has managed a team of chemists in Restek’s innovations laboratory who perform new product testing, method development, and applications work since 2004. Before taking the reins of the laboratory, he spent seven years as an environmental chemist and was critical to the development of Restek’s current line of volatile GC columns. Prior to joining Restek, he operated a variety of gas chromatographic detectors conducting method development and sample analysis. Chris holds a B.S. in environmental science from Saint Michael’s College, USA.
E-mail:chris.english@restek.com
Website:www.restek.com



















New Study Reviews Chromatography Methods for Flavonoid Analysis
April 21st 2025Flavonoids are widely used metabolites that carry out various functions in different industries, such as food and cosmetics. Detecting, separating, and quantifying them in fruit species can be a complicated process.

University of Rouen-Normandy Scientists Explore Eco-Friendly Sampling Approach for GC-HRMS
April 17th 2025Root exudates—substances secreted by living plant roots—are challenging to sample, as they are typically extracted using artificial devices and can vary widely in both quantity and composition across plant species.

