Increasing Proteome Coverage with ETD and CID in One Run Using Intelligent Data-Dependent Decision Tree Logic
Thermo Fisher Scientific Application Note
Martin Zeller, Torsten Ueckert, Bernard Delanghe and Thomas Moehring, Thermo Fisher Scientific, Bremen, Germany.
Since the addition of electron transfer dissociation (ETD) to the Thermo Scientific LTQ Orbitrap hybrid mass spectrometers, ETD has become a standard tool for proteomics research together with the traditional collision-induced dissociation (CID).1 Whereas CID produces b- and y-fragment ions, ETD predominantly produces c- and z-type fragment ions.
ETD has shown to be a complementary fragmentation technique to CID and Josh Coon and coworkers have found rules for on-the-fly decisions whether to use CID or ETD, depending on the peptide charge state z and mass-to-charge ratio (m/z) which more likely results in a confident peptide identification upon database search. These rules have been implemented in a data-dependent decision tree (DDDT) logic in the instrument method set-up.
A raw file acquired with the DDDT method contains therefore both CID and ETD MS–MS spectra, which require different search parameters for a database search and, in addition, ETD spectra require pre-processing before submission to the search engine.
The set-up of the DDDT method within the Thermo Scientific Xcalibur method editor is straightforward. In the standard DDDT method, all doubly charged peptides are fragmented with CID, for higher charged peptides CID or ETD is used depending on the peptide's m/z (see Table 1). For peptides with charge states higher than five ETD is used. In this work, we compare the results of the DDDT method with the results of CID and ETD only methods.


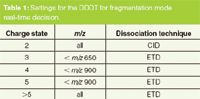
Table 1: Settings for the DDDT for fragmentation mode real-time decision.
Experimental
All spectra were acquired on the Thermo Scientific LTQ Orbitrap XL ETD. 500 ng of the complex Saccharomyces cerevisiae samples were separated by nano-LC in a 130 minute gradient. The LTQ Orbitrap XL ETD performed a high-resolution, accurate mass survey scan followed by eight data-dependent MS–MS scans with detection of the fragment ions in the linear ion trap.
Data analysis was done using Thermo Scientific Proteome Discoverer 1.1 software suite using Sequest, Mascot and ZCore. Peptide precursor mass tolerance was set to 10 ppm and fragment ion mass tolerance to 0.6 Da for CID and 1.1 Da for ETD (taking hydrogen re-arrangement for ETD fragment ions into account). Carbamidomethylation on cysteine residues was used as fixed modification and oxidation of methionine as variable modification. ETD spectra were pre-processed using the 'Non-Fragment Filter'.3,4 For the DDDT data, the MS–MS spectra were filtered for activation type and the appropriate search parameters for CID and ETD were used.
All spectra were searched against NCBI database filtered for yeast proteins containing 26918 entries using a target false discovery rate (FDR) of 1%.
Results
To make use of the complementary character of CID and ETD without the DDDT logic, samples would have to be injected twice (once for the CID run and once for the ETD run). This approach doubles instrument time as well as sample consumption.
Figure 1 illustrates the results of the two approaches using Venn diagrams for the distribution of identified peptides for the two runs in Figure 1(a) and the one DDDT run in Figure 1(b). For the two runs with the CID and ETD method almost twice (91%) as many MS–MS spectra have been triggered but only 16% more peptides and 10% more proteins are identified compared to the single DDDT run. The number of identified peptides using CID is almost the same for both approaches whereas the number of peptides identified by CID and ETD or by ETD only is less than for the DDDT run. The reason for this is twofold: 1) The sample was enzymatically degraded to rather short peptide stretches, which predominately ionized doubly charged peptides; 2) CID is superior for doubly charged peptides compared to ETD whereas ETD is more efficient for higher charged peptides. This behaviour underscores the power of the DDDT approach to choose the appropriate dissociation method that most likely result in confident peptide identification.

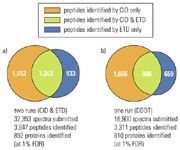
Figure 1: Venn diagrams of the identified peptides using CID and ETD method (a) and DDDT (b).
Conclusion
We have shown that the data-dependent decision tree method improves peptide and protein identifications compared to separate runs using CID and ETD. This is especially useful when low sample amounts and/or limited instrument time are available. Furthermore, we show that the Proteome Discoverer software has all the tools that are necessary for data mining of mixed raw files containing CID and ETD spectra.
Acknowledgements
We would like to thank Dr David M. Good (Karolinska Institute Stockholm, Sweden) for the kind gift of the yeast digests.
References
1. G.C. McAlister et al., J. Proteome Res., 7, 3127–3136 (2008).
2. D.L. Swaney, G.C. McAlister and J.J. Coon, Nat. Methods, 5, 959–964 (2008).
3. M. Zeller et al., Proceedings of the 57th ASMS Conference; Philadelphia, Pennsylvania, USA, June 2009, M150.
4. M. Zeller et al., Thermo Scientific Application Note 30179.
Mascot is a trademark of Matrix Science Inc. SEQUEST is a registered trademark of the University of Washington. LTQ Orbitrap, LTQ Orbitrap XL ETD, Xcalibur and Proteome Discoverer are the property of Thermo Fisher Scientific Inc. and its subsidiaries.


Thermo Fisher Scientific
Hanna-Kunath-Strasse 11, 28199 Bremen, Germany
Website: www.thermoscientific.com





















Removing Double-Stranded RNA Impurities Using Chromatography
April 8th 2025Researchers from Agency for Science, Technology and Research in Singapore recently published a review article exploring how chromatography can be used to remove double-stranded RNA impurities during mRNA therapeutics production.






