Implementing New Technology in a Regulated Environment
LCGC North America
After having collected some industry accepted practices for implementing new LC technology, the duo discuss FDA guidance available on the subject with respect to LC, and using some recently introduced LC technology.
In this column, we have collected some industry-accepted practices for implementing new LC technology, will discuss FDA guidance available on the subject with respect to LC, and using some recently introduced LC technology, will provide some examples on how to adopt this new technology for already approved, standard, or validated analytical methods.
Changes to an Approved Method
First, a disclaimer: If you decide to continue, what you are about to read is a summary of accepted practice from the authors' experience and in the form of both regulatory guidelines as well as the authors' opinions derived from a recent informal poll of industry and regulatory resources on the subject of a change in a validated LC method. We found out that regulating change is somewhat of a gray area, but there is guidance available. Please consult with the proper authorities before implementing any plan based on this article. Bottom line, as always, is that good, justifiable science is always the desired solution, and it is a good idea to have standard operating procedures (SOPs) in place to follow.
Standard or validated methods can be found in a number of places, for example, the United States Pharmacopeia (USP), or in the Official Methods of Analysis of the Association of Official Analytical Chemists (AOAC). Methods in both new (NDA) and abbreviated (ANDA) drug applications also are considered to be standard validated methods.
To use a standard method as is for the first time, a laboratory must perform a verification to demonstrate that both instrument and method performance criteria are met (1–3). However, to implement new technology, an adjustment, or a modification or change to a standard method might be needed. In April of 2004, the FDA published a guidance that makes recommendations to holders of both NDAs and ANDAs that desire to make postapproval changes (4). It is important that analysts refer to this guidance to determine what type of changes-being-effected supplement is recommended. In the guidance, the FDA provided reporting categories depending upon the type of change, or the potential to have an adverse effect on the identity, strength, quality, purity, or potency of a drug product.
A major change is a change that has a substantial potential to have an adverse effect. In the case of a major change, a Prior Approval Supplement (PAS) is required. As the name implies, FDA approval is necessary prior to distribution of the drug product made using the change. A moderate change has a moderate potential for an adverse effect, and requires the submission of a supplement called a Supplement-Changes Being Effected in 30 Days, often referred to as a CBE-30. For changes in this category, the drug product cannot be distributed if the FDA informs the applicant within 30 days that a PAS is required. CBE supplements also are used without the 30-day window for some moderate changes that do not relax acceptance criteria or for those that provide the same or increased assurance of identity, strength, quality, purity, or potency of a drug product. There have been cases where the FDA has not been able to complete a CBE-30 review within 30 days. If a firm were to implement the procedure after 30 days without FDA review, they are within the regulation, but at risk. There also have been cases where the FDA has notified firms that a review would not be completed within 30 days and has asked the firm not to implement the method. Each of these cases is an example of where it is important to have a good dialog with the FDA.
A minor change is a change that has minimal potential for an adverse effect, and these changes are described in the applicant's next annual report.
A supplemental or annual report must include a list of the detailed description for all of the changes; for supplements, the information is to be summarized in the cover letter, in annual reports, included in the summary section. Applicants also are encouraged to submit a comparability protocol that describes test, studies, and acceptance criteria used to demonstrate the absence of any adverse effects, and guidance on this topic recently has appeared in draft form (5). However, if a comparability protocol was not included in the original application, then it must be submitted as a PAS. The type of supplement also is dictated somewhat by the types of sample, for example, drug product versus intermediate.
What Constitutes a Change to a Method?
Adjustments to USP methods have been allowed to satisfy system suitability requirements as often noted in individual monographs. However, at what point does an adjustment become a change? After all, a change in the method triggers a revalidation and at least some level of reporting as outlined previously. Historically, as long as adjustments to the method are made within the boundaries of any robustness studies performed, no further actions seem to be warranted. However, any adjustment outside of the bounds of the robustness study constitutes a change to the method, requiring a revalidation.
In 1998, Furman and colleagues proposed a way to classify allowable adjustments (6). However, it was not until 2005 that guidance appeared on the topic (3,7,8). While USP guidance is still in revision (7,8), the FDA Office of Regulatory Affairs (ORA) has had guidance in place for a number of years (3), and Table I summarizes the adjustments allowed for various LC and gas chromatography (GC) parameters taken from both the USP and ORA documents. Adjustments outside of the ranges listed in Table I constitute modifications, or changes, which are subject to validation. While the criteria in Table I might seem quite straightforward, many do not account completely for recent advances in LC technology, especially columns with much smaller particle sizes, and some additional comments are warranted from an LC method perspective. Also, missing in the guidelines is a discussion on gradient adjustments and modifications. Although solvent composition itself is addressed, compensation for gradient delay volume and maintaining the same number of gradient column volumes when changing between different column dimensions also must be scaled properly for equivalent results. In addition, identical column chemistry, while not explicitly stated, must be implied.
Temperature and pH Adjustments
As shown in Table I, the pH value of the buffer in the mobile phase can be adjusted to ±0.2 pH units. Adjusting the pH should, however, take into account the pKs of the compounds of interest; because near the pK, even a 0.1 unit change in the pH can result in significant (>10%) changes in retention time (9). Studies show that for many compounds only operating at pH extremes (pH > 8 or pH < 4 for basic compounds, pH < 3 or pH > 7 for acidic compounds), generally well away from the compound pK, will accommodate the ±0.2 unit allowable change, due to the slope of the pH versus retention curve (10).
In the case of LC column temperature, where a ±20% change is allowed, significant selectivity effects might be encountered, where at 35 °C, a ±20% change would mean a ±7 °C difference. At higher temperatures somewhat common today for faster analyses, the change becomes even more significant.


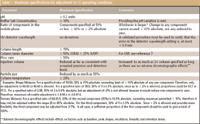
Table I: Maximum specifications for adjustment to LC operating conditions
Column Length, Diameter, and Particle Size Adjustments: Scaling the Separation
A few inconsistencies exist in guidelines regarding the flow rate and column internal diameter, length, and particle size adjustment criteria. It is possible to reduce flow rate and internal diameter (as much as 50% allowed by ORA, 25% by USP) farther than that listed in Table I as long as a constant linear velocity (given by the ratio of the squares of the internal diameter) is maintained, and still obtain equivalent results. Column length, internal diameter, and particle size adjustments really need to be considered together, and when correctly scaled according to well known theoretical principles, equivalent separations will result even outside the recommended adjustment criteria. For example, keeping the length to particle size ratio (L/dp) constant, an identical separation can be obtained for a 5-cm, 1.7-μm column as for a 30-cm, 10-μm column (L/dp = 3 for both) as long an increase in the flow rate inversely proportional to the particle size also is maintained.
By properly scaling other parameters as necessary, and by using instrumentation properly designed for these applications, a separation on the two columns (assuming the same chemistry) will be identical, even though the column length and particle size adjustment is a reduction of about 84%. A corresponding reduction in column diameter and flow rate of over 50% also would not impact the results.



Table II: Commercially available sub-2-μm particle columns
Let us look at an example of converting or migrating a method from high performance LC (HPLC) to newer LC technology that uses sub-2-μm particle size chemistry. Suppliers of chemistry (sub-2-μm particles) and instrumentation (systems capable of pressures greater than 10,000 psi) necessary to take advantage of this new technology are listed in Tables II and III, respectively (see also references 11–13).



Table III: Commercially available systems with 10,000 psi or greater capabilities
A few easy steps using equations that geometrically scale the original method to the new column packed with sub-2-μm particles using exactly the same mobile phase composition are necessary to achieve equivalent results. These equations take into account the changes in the gradient time (unless using isocratic conditions), flow rate, and injection volume.
The gradient is scaled using the following equation:


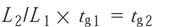
where L1 and L2 are the lengths of the original and new columns, and tg1 and tg2 are the times of each gradient step, respectively.
Flow rate is scaled taking into account the difference in the diameter of the two columns:


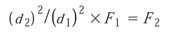
where d2 and d1 are the column diameters and F1 and F2 the flow rates.
To keep the column volumes proportional, the gradient steps should be readjusted for the new flow rate:


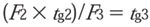
where F2 and tg2 are the flow rate and gradient time of the geometrically scaled values and F3 and tg3 are the optimized values. F3 is usually increased above that calculated for F2 (0.5 mL/min in the following example), to better approximate the optimum linear velocitiy for a sub-2-μm particle.
The injection volume is scaled taking into account the volumes of the two columns:


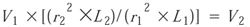
where r22 and r12 are the radii of the columns L1 and L2 are the lengths of the columns, and V1 and V2 are the injection volumes.
Laboratories might be interested in implementing this new technology to save time and expense compared to existing standard methods. Figure 1a shows an HPLC separation of a series of related caffeic acid derivatives from Echinacea purpurea, a natural product. When column reequilibration is taken into account, the run time exceeds 40 min. When properly scaled for injection volume, flow rate, and gradient time, the separation illustrated in Figure 1b is obtained. The run time is complete in under 6 min including reequilibration, increasing throughput approximately 7X, while using about 10X less solvent. All this without really changing the look of the separation; if it was not for the time scale scale in the figures, it would be difficult to distinguish between the two separations. But, to implement the method, we can be faced with a number of situations. Do we:



Figure 1
- Implement it as an existing standard method (USP, AOAC, or method in an approved NDA or ANDA)?
- Implement it as an existing standard method with adjustments?
- Implement it as an existing standard method with modifications or changes?
We will review each of these situations in turn and see how they apply to any given set of circumstances.
Implement an Existing Standard Method
To implement an existing standard method, or to determine the suitability of the method under actual conditions of use, verification is necessary to confirm that the method works for a particular drug substance, excipients, or dosage form by verifying a subset of validation characteristics, rather than completing a full validation, as proposed in a new USP chapter, 1226 (1,2). Chapter 1226 is considered an extension of Chapter 1225, and the intent is to provide guidance on how to verify that a compendial procedure that is being used for the first time will yield acceptable results utilizing the laboratories' personnel, equipment, and reagents. Verification consists of assessing selected Analytical Performance Characteristics described in Chapter 1225 to generate appropriate relevant data as opposed to repeating the entire validation process. Verification has been covered in some detail in a previous Validation Viewpoint column, and should be consulted for more information (1). Verification data and any statistical evaluation of equivalence should be included in the annual report.
Implement an Existing Standard Method with Adjustments
When implementing an existing standard method with adjustments the main thing to keep in mind is that as long as the adjustments are within the guidelines (Table I) or within the bounds of a robustness study, it is not necessary to perform a revalidation. According to the ORA guideline the modified procedure should not adversely affect the precision and accuracy of the data generated as measured against the performance specifications of the method.
Changes in column length, diameter, and particle size outlined previously to implement sub-2-μm LC, although outside of the criteria listed in Table I, when done properly really fit into this category, and guideline revisions are underway to accommodate this apparent discrepancy, at least for the particle size (7,8). This interpretation is only true, however, if the chemistry of both the stationary and mobile phase is nearly identical between the old and new conditions. Tools exist to approximate column stationary phase equivalence, and additional guidance has been proposed in this area (14,15). Adjustments in this category are usually accommodated in an annual report, but some companies, acting conservatively, can use a CBE or CBE-30 document. Actual determination of which document to file can be part of a risk assessment, or an existing SOP. And again, as mentioned previously, the type of supplement also is dictated somewhat by the types of sample, for example, drug product versus intermediate.
Implement an Existing Standard Method with Changes
When a new method is implemented in a regulated laboratory, it must be revalidated. Validation also is required when the existing standard method is modified enough to change it, and also a good idea when the existing method is applied to a sample matrix significantly different from that for which the original method was intended. There are many reasons to change a method, and changes to a method can be either reactive, or proactive. Reactive situations might exist if there were significant changes to incoming raw material or the manufactured batch, or formulation changes. If it becomes necessary to modify a method to satisfy system suitability requirements so much that it becomes a change, it also might be necessary to perform an out-of-specification (OOS) investigation (16,17).
Many laboratories are proactive with method changes, and implementing new technology fits into this category. New columns, column chemistry, and other method improvements occur frequently, and business case studies are undertaken to determine what changes might be made as cost cutting or time saving options. With run times as short as 1 min, with improved sensitivity and no loss in resolution, and software tools available to analyze the data more quickly, revalidation time is significantly reduced. Some will maintain that "if it ain't broke, don't fix it." But many companies take advantage of new technology because it makes sense from a business standpoint, even if it means changing the method, investing additional time and resources, and doing a little extra paperwork.
Changes themselves can be of different magnitudes, and result in different approaches for implementation. If the changes are so drastic that the applicant is essentially establishing a new or alternative analytical procedure, a PAS is required as this situation falls into the major change category. The same is true in the instance where a change is being made to relax specifications.
However, when adopting new LC technology, we generally operate under the assumption that a change is always being made for the better; that is we are not relaxing specifications but providing an increased assurance of identity, strength, purity or potency of the material being tested. It also can be further argued that adopting new LC technology is not equivalent to adopting an alternative procedure. A change in column chemistry or scale should not be rated the equivalent of changing from an HPLC method to a titration method, or to nuclear magnetic resonance (NMR) or IR spectroscopy, or vice versa. So a change in this category also might be satisfied by either type of CBE document. Of course, the standard method is the legal control procedure. Any change to the procedure would require a CBE-30 submission.
One final recommendation; when implementing a change, equivalency studies with the old method always should be undertaken to identify potential bias. Method equivalency is particularly important if the method is changed in-between points in a long term stability study. The applicant should provide information to explain why the new method is preferred to the original, including supporting data.
Conclusion
While it is certainly easier to adopt new technology with new methods rather than to revalidate current standard methods, implementing new technology can have a significant return on investment that can make a method modification a very worthwhile pursuit. By consulting the guidelines and adhering to a few basic principles, change can be implemented in a painless exercise that can result in faster, more sensitive, more robust information that might even reveal a wealth of new useful information.
Acknowledgments
The authors would like to acknowledge Uwe Neue, Eric Grumbach, Jeff Mazzeo, Diane Diehl, Tom Wheat (Waters Corporation), and numerous contributions from various industry leaders.
Michael E. Swartz "Validation Viewpoint" Co-Editor Michael E. Swartz is a Principal Scientist at Waters Corp., Milford, Massachusetts, and a member of LCGC's editorial advisory board.
Ira S. Krull "Validation Viewpoint" Co-Editor Ira S. Krull is an Associate Professor of chemistry at Northeastern University, Boston, Massachusetts, and a member of LCGC's editorial advisory board.
The columnists regret that time constraints prevent them from responding to individual reader queries. However, readers are welcome to submit specific questions and problems, which the columnists may address in future columns. Direct correspondence about this column to "Validation Viewpoint," LCGC, Woodbridge Corporate Plaza, 485 Route 1 South, Building F, First Floor, Iselin, NJ 08830, e-mail lcgcedit@lcgcmag.com
References
(1) M.E. Swartz and I.S. Krull, LCGC 23(10), 1100–1109 (2005).
(2) Pharmacopeial Forum, 31(2), 555 (Mar.-Apr. 2005).
(3) FDA ORA Laboratory Procedure, #ORA-LAB.5.4.5, Revised 09/09/2005. See also: http://www.fda.gov/ora/science_ref/lm/vol2/section/5_04_05.pdf
(4) Guidance for Industry, Changes to an Approved NDA or ANDA, FDA Center for Drug Evaluation and Research, Rockville MD, April 2004. See also: http://www.fda.gov/cder/guidance/index.htm
(5) Guidance for Industry, Comparability Protocols-Chemistry, Manufacturing, and Controls Information, FDA Center for Drug Evaluation and Research, Rockville, Maryland, February 2003. See also: http://www.fda.gov/cder/guidance/index.htm
(6) W.B. Furman, J.G. Dorsey, and L.R. Snyder, Pharm. Technol. 22(6), 58–64 (1998).
(7) Pharmacopeial Forum, 31(3), 825 (May-June 2005).
(8) Pharmacopeial Forum, 31(6), 1681 (Nov.-Dec 2005).
(9) M.E. Swartz, unpublished data on the analysis of tricyclic amines at pH 7.2.
(10) M.E. Swartz and I.S. Krull, LCGC 23(6), 46 (2005).
(11) M.E. Swartz, J. Liq. Chrom. & Rel. Technol. 28(7-8), 1253–1263 (2005).
(12) Separation Science Redefined, Supplement to LCGC, May 2005.
(13) J. Mazzeo, U. Neue, M. Kele, and R. Plumb, Anal. Chem. 277(23), 460A–467A (2005).
(14) Pharmacopeial Forum 31(2), 637 (March-April 2005).
(15) Pharmacopeial Forum 32(3), 833 (May-June 2006).
(16) M.E. Swartz and I.S. Krull, LCGC 23(6), 26 (2004).
(17) Guidance for Industry, Investigating Out of Specification (OOS) Test Results for Pharmaceutical Production (U.S. Food and Drug Administration, Department of Health and Human Services, Rockville, Maryland, September 1998)















University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

Prioritizing Non-Target Screening in LC–HRMS Environmental Sample Analysis
April 28th 2025When analyzing samples using liquid chromatography–high-resolution mass spectrometry, there are various ways the processes can be improved. Researchers created new methods for prioritizing these strategies.





