Spectral Interpretation, Part II: Tools of the Trade
LCGC North America
In Part I of this two-part look at spectral interpretation, Michael Balogh examined the ability to see and appreciate the product of our analytical devices. In Part II, he takes a closer look at the tools of the trade.
In Part I of this look at spectral interpretation (1), we examined our ability to see and appreciate the product of our analytical devices. In Part II, we examine the tools of the trade.
For many, software is the "Holy Grail." It is not because they lack the requisite expertise to interpret spectra. Indeed, many well-recognized experts rely on software tools. Rather, software can be expeditious, reducing huge volumes of data while highlighting issues the unaided eye might overlook. Software can help us reduce uncertainty if, with properly applied skills, we make use of the electron valence rules for nitrogen-containing compounds, characteristic spectra of halides, rings-and-double bond calculations, and so forth to arrive at what we believe is an unambiguous conclusion. Yet, if truth be told, no single software application can answer all inquiries satisfactorily. And so, what really counts is a practitioner's ability to apply well-honed skills and educated judgment.
There are two levels of access to interpreting mass spectra: nominal mass data and exact mass data. In each case, retention times serve as an additional determinant. Achieving accurate mass measure is based upon the calculated elemental composition. Not surprisingly, accurate isotope patterns fed into an algorithm to reduce the number of possible formula candidates is a recently exploited aspect of accurate mass measurement. Waters (Milford, Massachusetts) offers isotope characterization — iFit — as an integral part of MassLynx software to improve the use of accurate mass data as does Bruker Daltonics (Billerica, Massachusetts) with its Sigmafit. For nominal mass output third-party products such as Advanced Chemistry Development's (Toronto, Canada) recently introduced IntelliXtract software can work with all data. Finally, National Institute of Standards and Technology (NIST) maintains significantly helpful spectral libraries and search-and-match algorithms.
HighChem (Bratislava, Slovak Republic) also offers libraries. Distributed by Thermo Fisher, its Mass Frontier software includes, in addition to reference libraries of tandem spectra, collated fragmentation mechanisms from decomposition pathways published in peer-reviewed journals. For many years, HighChem has collected fragmentation mechanisms based upon mass spectrometry (MS) results published in printed media. According to the company's website (www.highchem.com), each reaction, together with chemical structures, is manually drawn in a graphic editor and saved in a library that includes some 19,000 individual reactions. Complementary information such as the title, authors, and source of the information accompanies the fragmentation pathways. This library collection serves as a knowledge base for predicting fragmentation pathways from user-provided structures.
It is noteworthy that previously reported mechanisms that Mass Frontier finds incorrect are corrected or excluded from the library. HighChem calls this feature automatic evaluation and says it consists of "simple element, charge, and radical consistency checks on both sides of the reaction, in addition to newly developed algorithms for complex electron mapping."
Recommended Reading
Regular readers of this column know that I focus my interest primarily on small-molecule work, and that I am involved with the Conference on Small Molecule Science (CoSMoS). Many of the topics appearing in this column are presented annually at CoSMoS. The utility of accurate mass measurement in its various applications is one many of us are spending an increasing amount of time on. As software evolves, accurate mass measurement is revolutionizing our practice. Time-of-flight (TOF) resolution, including that of hybrids like the quadrupole TOF (QTOF) system, can exceed conventional quadrupole resolution 10 over with mass accuracy falling within a few parts per million of the trur calculated monoisotopic or exact mass value. These characteristics make empirical formula determination possible based upon mass defect, where the critical mass value of H is a differentiator. Speciation, too, is more accurately accomplished. Consider a modest increase in mass accuracy beyond quadrupole limits to only 30 ppm. We can now discern between an aldehyde and a sulfide, where the two differ by 0.035 Da. Consider also differentiation between metabolic processes. In methylation (addition of CH2), TOF systems yield an increase in the measured mass over the response for the drug alone of +14.0157. Compare this with a two-stage biotransformation involving hydroxylation (addition of O2) followed by oxidation at a double bond (loss of H2) that yields an increase of +13.9792. Yet, significantly, when limited by the nominal resolution typical of the quadrupole response, both measurements will look like +14.
Instruments are becoming more utilitarian and egalitarian — their prices are coming down. Of the many entry-level discussions on the role isotopes play in determining a compound's identity, one appeared recently in LCGC Europe that contributes a helpful balance. "Interpretation of Isotope Peaks in Small Molecule LC–MS" (2) is based upon low-resolution ion trap work. In a relevant part, the author, Lionel M. Hill, cautions against overconfidence when using ion traps:
"[I]on trap users will have to be more careful than those with QTOF or triple quadrupole systems. It is obviously necessary to start with the +1 isotope peak isolated free of contamination . . . ion traps tend to trap with lower resolution than they scan . . . empty[ing] the trap . . . in order of mass."
This does not mean ion traps cannot be used but like all instruments, must be applied with an understanding of their abilities and their limitations.
Resolution is an instrument's ability to differentiate increasingly smaller differences in mass, usually represented as the mass divided by the width at 50% of the peak height: the full-width, half-height maximum (FWHM) method. Resolution can be 106 –109 (and beyond) with some dedicated Fourier-transform MS instruments, which allow you to study isotopic fine structure differences and isomers. In practice, resolution from 5000 to 10,000 measured for all peaks in the range achieves the best balance of precision and data acquisition speed (that is, improved results by obtaining an increased number of points across an acquired peak).
You might also find an "MS — The Practical Art" column from 2004 useful. In "Debating Resolution and Mass Accuracy" (3), I discuss these fundamental concepts in comparison of high-end instruments. As an aside, because the article compares high-end instruments only, I should note comments made to me by a well-respected researcher in instrument design, Brian Green (Waters Manchester, UK), who employs quadrupoles extensively in his life science interests.
Low-resolution quadrupole instruments also are used in extremely high mass accuracy measurements. As an example of this, consider proteins, whose masses are generally defined as "average" values when the isotope peaks are not resolved from one another. Average mass is the weighted mean of all the isotopic species in the molecule. Adopting the resolution normally employed on quadrupole instruments, a 10-kDa protein broadens by a factor of 1.27. The broadening increases significantly as the mass increases (that is to ×2.65 at 100 kDa). However, reducing the calibrated peak width to a 0.25 mass-to-charge ratio (m/z) — about half the usual quadrupole-calibrated peak width when used for nominal mass measurement — increases the resolution to 4000, which improves things dramatically. When these ions are observed on an instrument set for a significantly lower resolution than that required to resolve the isotopes (say, less than 10,000 resolution), a single peak is produced for each charge state. For example, a 20-kDa protein with 10 or 20 charges on it produces isotope envelopes that are 0.9 m/z units wide at m/z ~2000 or 0.45 units wide at m/z ~1000.
Two papers for further reading on this topic were recommended by Green. In addition to showing the accuracy and precision achievable, the first paper (4) thoroughly covers the basics and contains references to papers that cover the historical background. The second (5) is a follow-up to the first. The main difference in the second instance is the authors coupled liquid chromatography (LC) to MS. Both papers refer to measurements made on the lowest isotopic species (monoisotopic species) of low molecular weight molecules, which is the usual application of exact mass measurement. The masses of the isotopes are extremely accurately known (relative uncertainty <1 in 108 ). Consequently, the monoisotopic mass of a compound can be very accurately defined — indeed to accuracy much better than can be achieved currently.
Completing the trilogy is an article by Oliver Fiehn and Tobias Kind, well-recognized names in the field of plant metabolomics, which was recently made available as a provisional web-based publication (6). In it, the authors characterize various database and search possibilities applied in highly complex mixture deconvolution, and they caution that placing too great a trust in accurate mass measurement alone invites error.
The First Step in Interpretation
In Part I of this series (1), we established that the starting point for structure determination comes from knowledge of a compound's molecular weight. Assessing molecular weight for the important components within a sample is always the first challenge, a function of sample complexity and the relationship of some or all the individual component peaks to the chemical entity examined. Thus, structure determination by mass spectrometry is ultimately a mixture of approaches that include determining an unknown's molecular weight, its fragment ions, and its neutral losses.
Today, despite the explosion of technological innovations that always have seemed to characterize our field, presenting data for examination still challenges the talents of many researchers. For example, NIST (National Institute of Standards and Technology, Gaithersburg, Maryland) developed AMDIS (Automated Mass Spectral Deconvolution and Identification System) for extracting and deconvoluting gas chromatography–electron ionization (GC–EI) data. But AMDIS is of limited applicability to interpretation of unknown components acquired by atmospheric ionization LC–MS. Component extraction and deconvolution approaches including component detection algorithm (CODA) (7) and matched filtration with experimental noise determination (MEND) (8) find applicability in LC–MS: each can reduce unwelcome solvent and background noise contributions and — we would hope — extract low-level chromatographic peaks. Yet, in practice, these broad-stroke approaches can sometimes obscure or remove important peaks of interest.
Nominal Mass Tools
About mass accuracy, Mark Bayliss, Director of Analytical Informatics at Advanced Chemistry Development (ACD/Labs), has this to say:
"Ideally, in all cases, we would have access to instrumentation that could provide five-decimal-place mass accuracy. However, even single quadrupole technology can be appropriate for some levels of structure interpretation. The challenge is to extract the maximum amount of information from the data we have at our disposal."
Relatively simple software tools can differentiate slow-moving solvent noise responses from the masses within a dataset that are eluted with a chromatographic peak-like response. The major disadvantage to these tools, as Bayliss points out, is that they stop short of component identification — they identify single-ion chromatogram peaks. But they do not show which ion chromatogram peaks combine to form a component or how they relate to each other. So, they can help show where in the dataset to look, which is useful when identifying trace compounds, but they do not completely remove the molecular weight identification bottleneck.
Bayliss says that his company's automated method for molecular ion determination, ACD/IntelliXtract (Figure 1), takes component extraction a step further. It analyzes the individual masses contributing to each chromatographic peak and determines which single-ion chromatogram peaks combine to form a true component. Then the software assigns the molecular ion for each component in the data, considering commonly associated process attributes such as adduct and multimer ions and 12 C and 13 C isotope ratios.


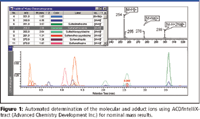
Figure 1
Once the molecular ions are identified, information that is useful for structure elucidation remains available in the spectrum "below" them. With software like ACD/MS Manager and HighChem Mass Frontier, you supply a chemical structure, predict fragments, and assign the fragments to the experimental spectrum. The software then predicts fragments according to a set of fragmentation rules and assigns the predicted fragments to the experimental spectrum. The major disadvantage is that you must suggest a possible structure before performing the assignment. Ultimately, even information-rich spectra generated by electron ionization suffer from anomalies introduced by background noise. De novo structure identification without any supporting knowledge of the structure requires a different approach, one that applies a combination of MS and, typically, nuclear magnetic resonance (NMR) spectroscopy.
Predicting useful sensitivity limits is sometimes difficult unless someone already has performed the extensive work required and published the results. A paper developed for pesticide analysis contains instructional data based upon AMDIS but is equally valid for all library search interests. For example, because noise is ubiquitous, establishing minimum sample concentrations is important inasmuch as it cannot be done by inspection. Spiked concentrations at 10 ng/mL produced no results (the instrument response was insufficient), while increasing the concentration to 50 ng/mL allowed AMDIS to extract and present positive results for nearly all test analytes, even though the target peaks were still not visible (9). GC (whether nominal mass or accurate mass) is of broad interest in mass spectrometry practice whether it be pharmaceutical, fine chemical, or environmentally applied.
Isotopes and Exact Mass
Accurate mass techniques relying on high fidelity characterization of isotopes associated with a particular spectrum provide an almost flawless response over and above that typically achieved by accurate mass response, which itself might differ from the exact mass of the entity only by a few parts per million.
Because of well-characterized datasets developed by TOF instruments enhanced by modern chromatography (ultrahigh-pressure, for instance), GC-like peak widths increase the homogeneity of a given peak, and fewer artifacts are included in the data presented to a search engine. Coupled to TOF-based instruments, thousands of potential candidates can be eliminated quickly from complex mixtures. Here, as in the GC example cited earlier, the instrument and its software work in a highly synergistic fashion. As Kind and Fiehn (6) note in their review of accurate mass results, elemental compositions cannot be determined unambiguously by mass accuracy alone, even at the exacting precision of less than 1 ppm. As noted in Part I, studies show most modern instruments used for accurate mass work achieve 10 ppm (1), and few if any can routinely achieve an accuracy of 1 ppm.
The answer is a combination of high-fidelity spectra and postrun isotope-based interrogation algorithms. Work in this area relies on well-characterized studies (11,12). Early efforts at isotope abundance calculations were predicted through polynomial means or, more recently, by fast Fourier transformations. Figure 2 shows the benefit of acquiring accurate mass measurements of not only the parent peak but the associated isotopes. Typically such software allows you to make informed choices of which filters to invoke, such as odd or even electron valences and minimum and maximum H/C ratios, and whether to weight the results with rings-and-double-bonds calculations.

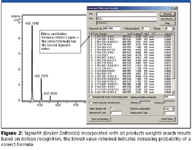
Figure 2
Recently, isotope and high mass accuracy filtering has been applied successfully in the most difficult application areas such as metabolomics and metabonomics studies and metabolism identification. In the latter, known metabolites, as well as novel or unknown ones, abound. The graph in Figure 3, from a bile study of the drug verapamil, indicates the unmanageable dilemma posed when no filters or controls are available, a common situation in metabolism studies because many of the unexpected results have no standards available for reference. Isotopes, as well as mass defect filters, increase the certainty of the search results amid impractical odds. The true atomic mass for the hydrogen (1.007825 as opposed to the nominal value of 1) is associated with the molecule and its fragments, whether displayed (charged) or lost as neutrals in the experiment. Without controls, 1169 possible peaks were detected, with no way to easily determine the drug-related compounds from endogenous entities. A control sample and nominal mass results reduce the number to a still-large list of 499 candidates. Controls and exact mass filters reduce the number to a far more manageable 26. Figure 4 shows simply how a mass defect filter works. The number of results displayed from among those in the entire data set is reduced by factoring out those lying outside the window for differences in mass from the theoretical of 40 mDa (in the Phase I example) or 70 mDa in the Phase II example. Figure 5 shows a similar study, in urine, in which 136 unexpected, possibly drug-related candidates were reduced to 39 legitimate compounds (13) by applying further filters specific to metabolic identification practice.

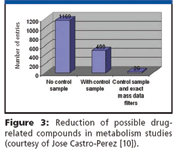
Figure 3
One dataset in the paper by Kind and Fiehn (6) is particularly striking, and it led to their conclusion based upon examination of 1.6 million formula search results:


Figure 4
"High mass accuracy (1 ppm) and high resolving power alone [are] not sufficient . . . only an isotopic abundance pattern filter [is] able to reduce the number of molecular formula candidates."

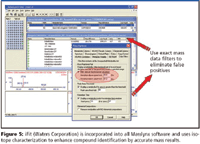
Figure 5
Mass spectrometers capable of just 3 ppm mass accuracy, but 2% isotope pattern accuracy, usually remove more than 95% of the false candidates. This performance would best even mass spectrometers capable of 0.1 ppm — if they actually existed — that are not equipped with isotope pattern capability. Between masses of 150 Da and 900 Da, the number of possible formulas listed as mass accuracy increased from 10 ppm to 0.1 ppm without the aid of isotope abundance information: from a low of 2 candidate formulas at 150 Da to 3447 at 900 Da for 10 ppm. Even at the upper end (900 Da) mass accuracy alone at 1 ppm yields 345 candidates. Invoking 2% isotope abundance accuracy, the number of candidates at 900 Da is reduced to an expedient 18. They also show that allowing a paltry 5% accuracy for isotope acquisition associated with 5 ppm accuracy yields 196 candidates. Indeed, the authors seem adamant when they aver that "Software producers should be enforced to use [isotopic abundance pattern] in their formula generation software . . ."
Software programs now, in addition to isotope ratio examination, include such comparators as detail derived from repeated sequential MS fragmentation experiments (HighChem Mass Frontier) or high–low energy collision fragmentation (14) to build relationship diagrams. When coupled with data mining programs such as principal component analysis software (for example, PCA SIMCA from Umetrics, Sweden) we can develop relational plots of what appear to be widely scattered data points. Therefore, as multivariate statistics becomes a necessary adjunct of the practice it was included this year as a CoSMoS workshop.
Clearly, as we discussed in Part I, operators will always need to bring their skills to bear when making the final decisions on what the data represents. Even with targeted analyses, the possibility always exists for novel chemistry to occur on the way to your results.
Some of the facets I omitted this time, or gave short shrift will surface again, because the topic of spectral interpretation is integral to MS practice. A note to the readers of this column: my thanks as always for your suggestions (and corrections). Your comments are quite valuable and often dictate which subjects appear here. Also, thanks to the authors of the cited publications for their invaluable contributions to our understanding, and to Mark Bayliss (Advanced Chemistry Development), Catherine Stacey (Bruker Daltonics), and Brian Green, Hilary Major, and Jose Castro-Perez (Waters Corporation) for their insights.
Michael P. Balogh "MS — The Practical Art" Editor Michael P. Balogh is principal scientist, LC–MS technology development, at Waters Corp. (Milford, Massachusetts); an adjunct professor and visiting scientist at Roger Williams University (Bristol, Rhode Island); and a member of LCGC's editorial advisory board.
References
(1) M.P. Balogh, LCGC 24(6), 580–587 (2006).
(2) L.M. Hill, LCGC Europe 19(4), 226–238 (2006).
(3) M.P. Balogh, LCGC 22(2), 118–130 (2004).
(4) A.N. Tyler, E. Clayton, and B.N. Green, Anal. Chem. 68, 3561–3569 (1996).
(5) T. Storm, C. Hartig, T. Reemtsma, and M. Jekel, Anal. Chem. 73, 589–595 (2001).
(6) T. Kind and O. Fiehn, BMC Bioinformatics 7, 234 (2006).
(7) W. Windig, J.M. Phalp, and A.W. Payne, Anal. Chem. 68, 3602–3606 (1996).
(8) V.P. Andreev, T. Rejtar, H.-S. Chen, E.V. Moskovets, A.R. Ivanov, and B.L. Karger, Anal. Chem. 75, 6314–6326 (2003).
(9) W. Zhang, P. Wu, and C. Li, Rapid Comm. in Mass Spectrom. 20, 1563–1568 (2006).
(10) J. Castro-Perez, "A new HTS UPLCTM–TOF-MS approach for in-vitro/in-vivo metabolite detection and identification," presented at Pittcon 2006.
(11) J.A. Yergey, Int. J Mass Spectrom. Ion Physics 52, 337–349 (1983).
(12) A.L. Rockwood, S. van Orden, and R. Smith, Anal Chem. 68, 2027–2030 (1996).
(13) R.J. Mortishire-Smith, D. O'Connor, J.M. Castro-Perez, and J. Kirby, Rapid Commun. Mass Spectrom 19, 2659–2670 (2005).
(14) M. Wrona, T. Mauriala, K.P. Bateman, R.J. Mortishire-Smith, and D. O'Connor, Rapid Commun. Mass Spectrom. 19, 2597–2602 (2005).










. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")

New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.


, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

