High-Throughput Peptide Mapping of Monoclonal Antibodies Using Tandem Liquid Chromatography– Mass Spectrometry
Analytical methods for quality control (QC) of monoclonal antibodies (mAbs) rely on peptide mapping workflows with liquid chromatography–mass spectrometry (LC–MS) systems. Whilst LC–MS is effective, peptide mapping with LC–MS can involve long analysis times that reduce the throughput of QC testing. Modern tandem LC methods can increase analysis efficiency and are designed to reduce the downtime of MS sampling to increase the efficiency of QC pipelines. The approach presented here improves the efficiency of mAb analysis peptide mapping, whilst returning high-quality data.
The development and production of therapeutics, such as monoclonal antibodies (mAbs), require high-throughput sample analysis and high‑quality data. For example, post-translational modifications (PTMs) can have a major impact on the quality and efficacy of the final product and, therefore, need to be monitored. Orthogonal analyses of protein PTMs are required to ensure certain critical quality attributes (CQAs) are within the established ranges to maintain lot-to-lot consistency in line with regulatory requirements and International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines (1).
Peptide mapping is considered a gold standard for mAb analysis because it delivers information on many attributes at the peptide level within a single analysis, providing a strong method for an analytical control strategy. One type of peptide mapping approach using liquid chromatography–mass spectrometry (LC–MS) analysis is the multi-attribute method (MAM), which allows high-quality, confident PTM identification in research and at later stages in quality control (QC) laboratories (2–4). However, peptide mapping with LC–MS methods can involve long analysis times due to the lengthy LC gradient needed to resolve peptides in the complex sample resulting from tryptic digestion. This creates challenges for high-throughput laboratories where efficiency is key. Moreover, long downtimes are also created between analyses to allow for column washing and re-equilibration steps, and when eluents are diverted to waste. During these periods, no separation is taking place. This reduces the time available to perform MS testing and reduces efficiency in research and QC pipelines.
Innovations in ultrahigh-performance liquid chromatography (UHPLC) instrumentation have led to the development of a tandem LC system that can be leveraged for high-throughput “tandem-mode” LC–MS analysis.
This article demonstrates the applicability of a tandem LC–MS workflow for peptide mapping analysis of trastuzumab in a high-throughput manner. With a tandem LC–MS approach, a greater number of sample injections were performed in the same timeframe, increasing analytical throughput and significantly reducing MS downtime. Chromatographic results were found to be equivalent across the two chromatographic channels. Additionally, the reproducibility of the obtained PTM values during a continuous 16-h period of operation were tested, demonstrating the stability of MS performance under continuous acquisition.
Materials and Methods
Sample Preparation: Samples were prepared following a published method (5) to obtain peptides from tryptic digestion of trastuzumab. Trastuzumab samples were diluted to 2 mg/mL in water. For each sample digest, sample, digestion buffer (buffer 1, pH 6.5 or buffer 2, pH 7.2), and 5 mM TCEP (final concentration) were added to each lane of a Thermo Scientific KingFisher deep well 96-well plate. Trypsin bead “wash buffer” was prepared by diluting digestion buffer 1:4 (v/v) in water. Bead buffer was neat digestion buffer. Digestion was performed using a Thermo Scientific KingFisher Duo Prime Purification System with Thermo Scientific BindIt software (version 4.0). Samples were incubated for 5 to 40 min at 70 °C on medium mixing speed to prevent sedimentation of beads for the digestion time course study, and beads were removed at each time point. Following digestion, 100-μL samples were transferred to 300‑μL Thermo Scientific PP Screw vials and cap and 1 μL of 10% trifluoroacetic acid (TFA) was added (final concentration 0.1% TFA) and immediately analyzed by high-resolution accurate mass LC–MS.
Chromatography Step: The resulted peptides were separated on a 2.1 × 250 mm, 2.2‑μm Acclaim Vanquish C18 column (Thermo Scientific) using the Thermo Scientific Vanquish Horizon Duo UHPLC System for tandem LC–MS workflows. Analysis was performed using a binary gradient of 0.1% (v/v) formic acid in water (A) and 0.1% (v/v) formic acid in acetonitrile (B) with a flow rate of 0.3 mL/min. The gradient started at 2% B and increased to 40% B over 45 min on the analytical pump, while the reconditioning pump performed two cycles from 2% to 80% B in 20 min and equilibrated back to 2% B for 25 min.
Mass spectrometry data acquisition was performed with the Thermo Scientific Orbitrap Exploris 240 MS system with biopharma option at resolution 120,000 FWHM. Application‑specific MS tune and acquisition settings are templated and provided within the Thermo Scientific Chromeleon Chromatography Data System (CDS) software, which makes this approach directly and easily transferable from instrument to instrument.
Tune Settings: Spray voltage: 3.8 kV; sheath gas: 25; auxiliary gas: 10; ion transfer tube temperature: 320 °C; vaporizer temperature: 150 °C; pressure mode: peptide.
MS Scan Settings: Resolution: 120,000 (at m/z 200); scan range: 200–2000 m/z; RF lens (%): 60; AGC target: 3 × 106; max injection time: 100 ms.
MS/MS Settings: Resolution: 15,000; normalized collision energy: 30; injection time: 200 ms; AGC target: 1 × 106; TopN: 5; microscan: 1; isolation window: 2 m/z.
Data were processed using the Thermo Scientific BioPharma Finder Software 4.0.
Results and Discussion
Peptide mapping is a routine analysis in biopharmaceutical assessment and is gaining more interest as part of the MAM workflow. It is driving the wider adoption of MS methods for later stages of biopharmaceutical product development and, ultimately, allowing its use in the QC laboratory. Method transfer onto the QC stage requires improved robustness and high‑throughput due to the high number of samples that must be processed and the required reliability of the methods assuring product quality.
In this study, trastuzumab monoclonal antibody was digested using a trypsin protein digestion kit and the generation of tryptic peptides was performed automatically on the purification system. The digested monoclonal antibody was then analyzed via tandem
LC–MS/MS analysis. CDS software automatically split the gradient across the two columns and pumps, managing when the analytical gradient was complete and the wash step started. Twenty‑one sample injections were performed in this study, alternating between the two columns to obtain a perfect overlap of runs coming from the two channels (Figure 1). This qualitative evaluation demonstrates the reproducibility of LC performance not only across the two channels but also when using two different columns.


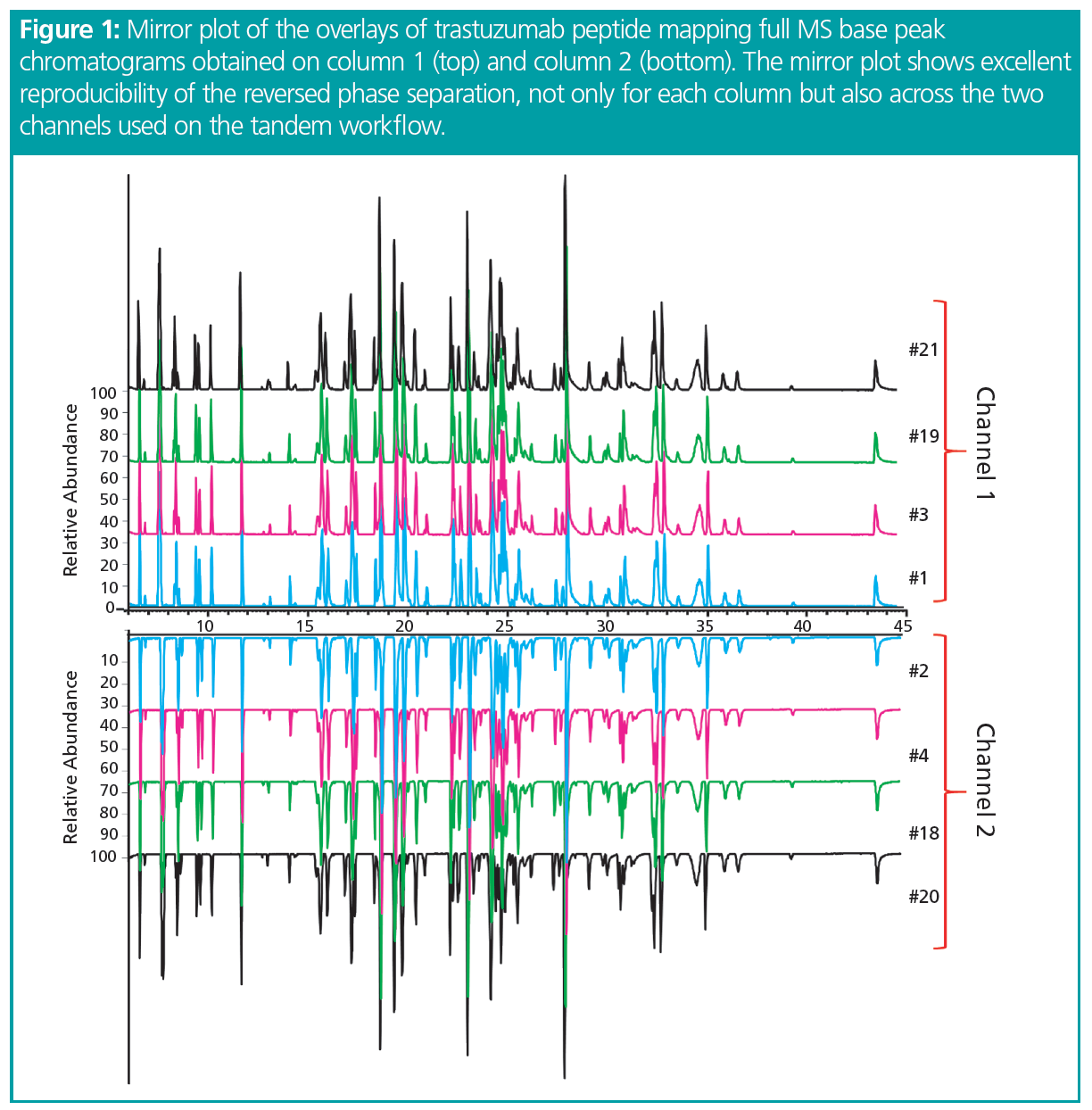
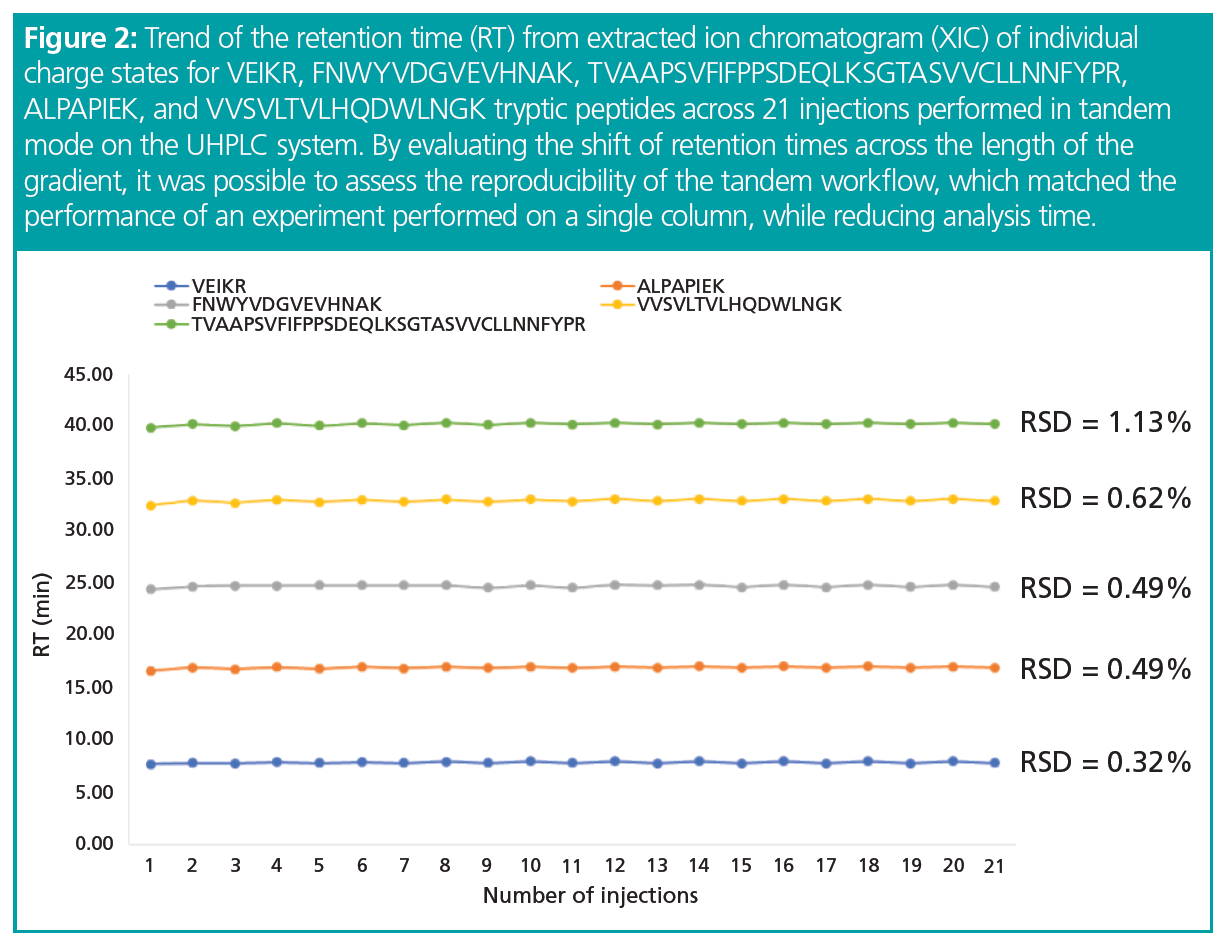
Chromatographic reproducibility was evaluated across the gradient by plotting the retention times for five different peptides along with the 21 injections. No difference was observed between the two data sets, returning overall relative standard deviation (RSD) values lower than 1.5% (Figure 2). Excellent retention time reproducibility is important for correct identification of components to evaluate and quantify drug product CQAs using MS. However, it would be possible to allow higher values of %RSD for the retention times when using high-resolution MS, as this level of resolution gives a confident identification of components even when using a wider retention time window.

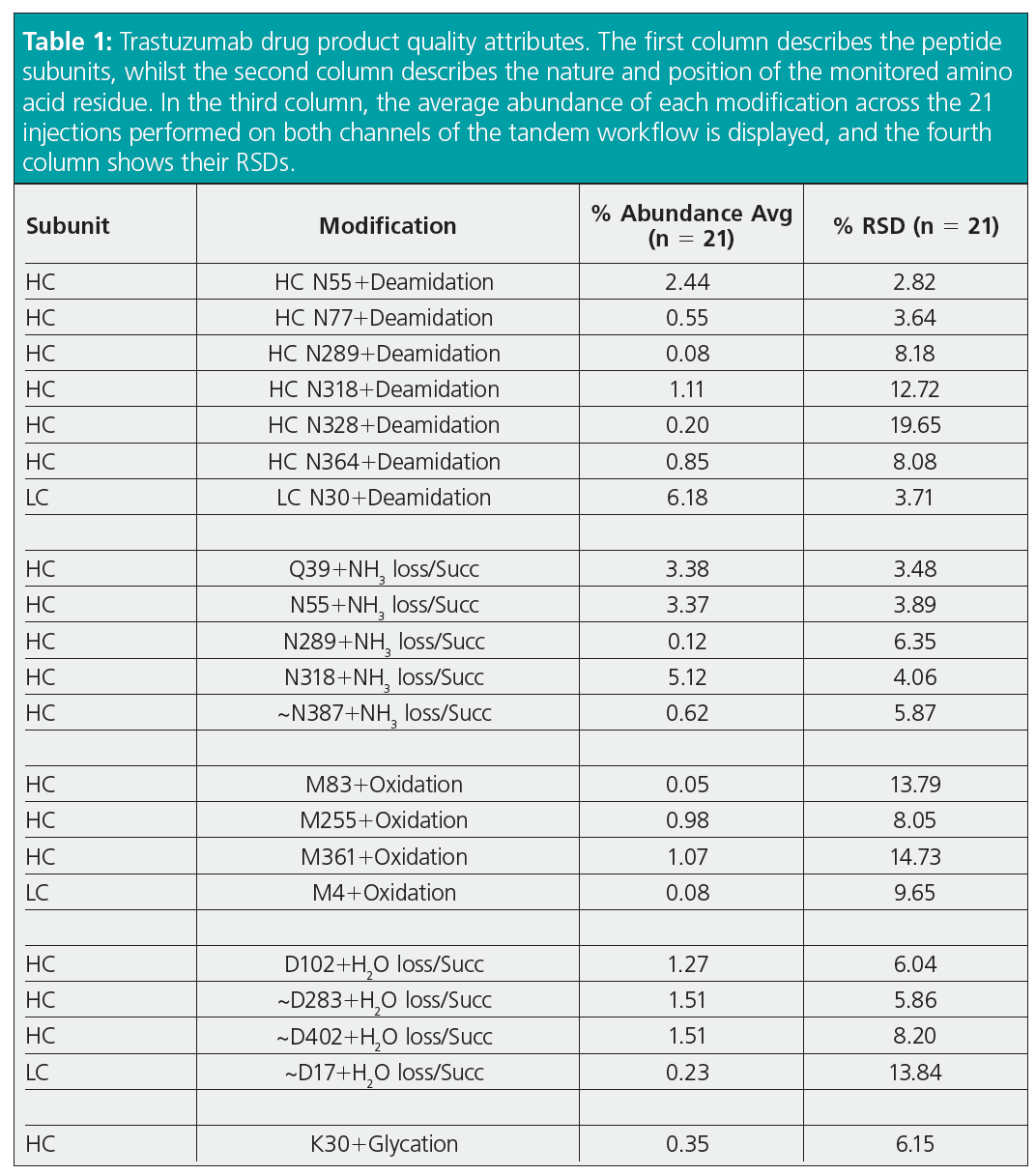
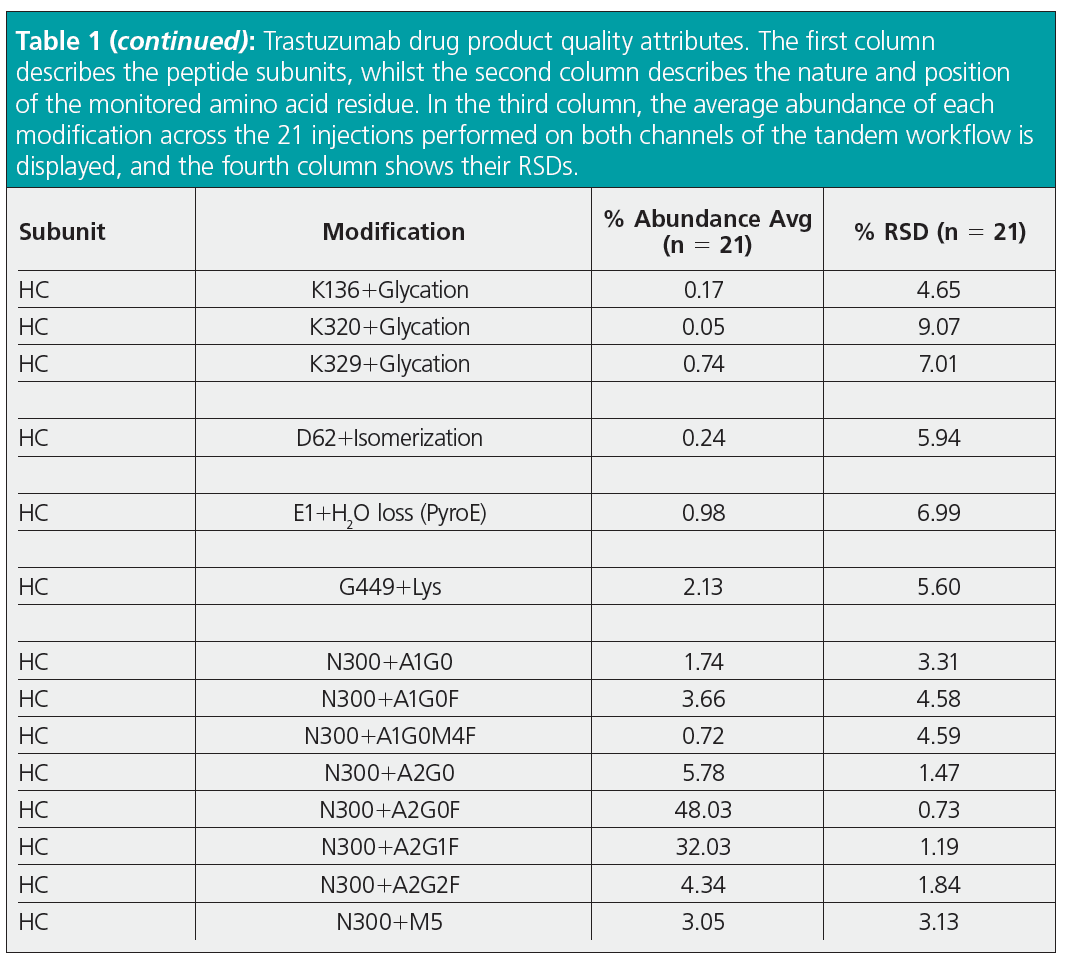
Peptide mapping analysis is mainly performed to evaluate PTMs in therapeutic proteins that need to be monitored during bioprocessing and batch release to guarantee drug product quality. For this reason, we evaluated product quality attributes for trastuzumab across the 21 injections performed in tandem mode (Table 1). Each modification was calculated considering up to one missed cleavage, and without considering Na+ and K+ adducts or non-specific cleavages, using a confidence score ≥ 95% and within ± 5 ppm. Detection of most PTMs resulted in very low RSD values, which exceeded 15% only for some low abundant modifications (N328 + deamidation = 0.20%). This proved excellent chromatographic equivalence of the two channels for PTM evaluation, as well as the suitability of the tandem LC–MS workflow for PTMs assessment and quantitation.


Conclusion
In this study, we have demonstrated the suitability of a tandem LC system for tandem LC–MS analysis for peptide mapping and the MAM workflow. This tandem LC approach significantly increased analytical throughput by reducing the time when MS was not fully utilized for valuable data generation; this was achieved by minimizing the time window where LC was diverted to waste and the MS not utilized. This workflow proved to be a more time-efficient alternative to traditional methods, while also maintaining or improving the performance and reproducibility required for a method to be considered for adoption in a QC laboratory.
Twenty-one samples of trastuzumab tryptic digest were analyzed using LC–MS/MS analysis in tandem mode. System performance was evaluated by monitoring retention time reproducibility and identifying PTMs, with both evaluations yielding excellent data and low %RSD values. Tandem analysis of peptide mapping replicates did not affect the accuracy of the results and the correct identification, but it had the benefit of saving at least 6 h of instrument time when compared to single mode during 24 h. The use of automated digest kits versus in-solution digestion has been shown to save between 2–20 h preparation time for each sample (6), providing significant sample preparation time savings, which, when combined with the efficiency benefits of the tandem analysis, can dramatically increase throughput.
In conclusion, tandem LC–MS represents a powerful way to increase laboratory productivity without compromising data quality, especially when combined with automated sample preparation of the tryptic digest. The operational simplicity of tandem LC–MS methods means that little training is required for high-throughput peptide mapping of biopharmaceuticals, enabling straightforward compliance-ready enterprise acquisition of LC–MS data for current good manufacturing practice (cGMP) environments.
References
- FDA-CDER-CBER, Guidance for Industry: Quality Considerations in Demonstrating Biosimilarity of a Therapeutic Protein Product to a Reference Product (April 2015).
- S. Rogstad, A. Faustino, A. Ruth, D. Keire, M. Boyne, and J. Park, J. Am. Soc. Mass Spectrom. 28, 786–94 (2017).
- R.S. Rogers, N.S. Nightlinger, B. Livingston, P. Campbell, R. Bailey, and A. Balland, Mabs 7, 881–90 (2015).
- R.S. Rogers, M. Abernathy, D.D. Richardson, J.C. Rouse, J.B. Sperry, P. Swann, et al., The APPS Journal 20, 1–8 (2018).
- S. Millán-Martín, C. Jakes, S. Carillo, et al., Anal. Bioanal. Chem. 412, 6833–6848 (2020).
- Thermo Scientific Application Note 72141, SMART Digest compared to classic in-solution digestion of rituximab for in-depth peptide mapping characterization, http://tools.thermofisher.com/content/sfs/brochures/AN-1159-SP-SMART-Digest-Peptides-AN72141-EN.pdf
Jonathan Bones obtained his B.Sc. in analytical science (chemistry) and Ph.D. in analytical chemistry from Dublin City University (DCU). He joined Pauline M. Rudd’s group at NIBRT in 2007. In 2010, Jonathan was appointed as the “John Hatsopoulos Research Scholarship” at the Barnett Institute of Chemical and Biological Analysis at Northeastern University in Boston, Massachusetts, USA, working under the mentorship of Barry L. Karger and investigating new approaches for glycomics and the application of proteomics for a better understanding of mammalian cell bioprocessing. In 2012, he returned to NIBRT.
Sara Carillo completed her Ph.D. in chemical sciences in 2013 at the University of Naples “Federico II”. Under the guidance of Professor Corsaro, she focused on the structural characterization of polysaccharides and glyco-conjugates from Gram-negative bacteria via nuclear magnetic resonance (NMR) and mass spectrometry techniques, focusing on the immunological properties and potentials of extremophiles endotoxins. After a period at the University College of Dublin, she joined Jonathan Bones’s research group in NIBRT in 2015, working on the understanding of the effects of extractables and leachables from single‑use bioreactors on CHO cells N-glycome and produced monoclonal antibodies. She is now working at NIBRT as Applications Development Team Leader for the development of new analytical approaches in biopharma.
Silvia Millán Martin is Applications Scientist in the Characterization and Comparability Laboratory in NIBRT, Ireland. She obtained her degree in pharmacy and Ph.D. in analytical chemistry from the University of the Basque country in North Spain, where her work focused on the development of LC–MS/MS- and GC–MS-based analytical techniques for the identification and quantitation of environmental, pharmaceutical, and nutritional significant compounds. She also worked for four years in a small contract research organization in the analytical services unit, and focused on the determination of bioactive compounds in natural sources and the application of metabolomics/proteomics approaches with the aim of evaluating their health benefits to potentially be used as functional ingredients in the food industry. In September 2012, she joined NIBRT, becoming part of the GlycoScience Group as a postdoctoral researcher in the mass spec lab and working on academic and contract research projects with biopharma companies. Her work focused on full detailed glycan analysis by MS and MS/MS and other orthogonal techniques, and on the development of advanced LC–MS‑based platforms for quantitative glycomics, with applications in bioprocessing and clinical biomarker discovery. In September 2016, she joined the Characterization and Comparability Laboratory, which is led by Jonathan Bones and collaborates with Thermo Fisher Scientific, to focus on the development of new applications for the characterization of biopharmaceuticals, and the associated processes used for their production.
E-mail: jonathan.bones@nibrt.ie / sara.carillo@nibrt.ie / silvia.millanmartin@nibrt.ie
Website: www.nibrt.ie


























New Method Explored for the Detection of CECs in Crops Irrigated with Contaminated Water
April 30th 2025This new study presents a validated QuEChERS–LC-MS/MS method for detecting eight persistent, mobile, and toxic substances in escarole, tomatoes, and tomato leaves irrigated with contaminated water.
, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.

University of Tasmania Researchers Explore Haloacetic Acid Determiniation in Water with capLC–MS
April 29th 2025Haloacetic acid detection has become important when analyzing drinking and swimming pool water. University of Tasmania researchers have begun applying capillary liquid chromatography as a means of detecting these substances.

