Efficient HPLC Method Development and Personal Reflections
LCGC Europe
A pioneer in high performance liquid chromatography reflects on his career and how "enlightened trial-and-error" can reduce the effort involved in method development.
Choosing conditions for a final separation ("method development") is often carried out by trial-and-error. With some experience and basic high performance liquid chromatography (HPLC) knowledge, this process can evolve into enlightened trial-and-error — in which fewer trials are required. Efficient method development aims at optimal results with minimal effort; it requires a quantitative understanding of how different experimental conditions affect the quality of a separation, and is best implemented with computer simulation. My own career, which emphasized improvements in method development, provides a background for the following short review.
My Path to Chromatography (1931–1956)
I was born in Sacramento, California, where in 1939 I received a chemistry set for Christmas. What proved fascinating was that stuff called compounds could be predictably assembled from something called atoms — nicely illustrated in the form of jigsaw cutouts (Figure 1).


Figure 1: An example of the jigsaw cutouts illustrating the assembly of atoms.
The concepts of valence and balanced equations were thus revealed, if in primitive form. This initial interest in chemistry grew over time with visits to the local library and later experiments (many dangerous!) in a home laboratory. When I entered the University of California at Berkeley in 1948, I was firmly committed to a career in chemistry.
An important refinement of this goal came during graduate research at Berkeley under Professor Donald Noyce (1952–1954). My research topic was the mechanism of an aldol condensation (1), which introduced me to physical-organic chemistry and model building. This provided an important tool for the next six decades ... the only thing missing was a research area in which to apply this tool. That would soon be supplied.
My first job was at Shell Oil in Houston, under the direction of M.J. ("Jack") O'Neal. Jack received the ACS Award in Petroleum Chemistry in 1956 for his pioneering development of mass spectrometry (MS) for the analysis of high-molecular-weight petroleum samples. Al Zlatkis, a future "impresario of chromatography," was part of the group when I arrived. A little later we were given the job of constructing a gas chromatography (GC) system using information from the proofs for a book by Keulemans (my first exposure to chromatography).
We began measuring retention times for various hydrocarbons, which led me to a connection between chromatography and model building in the form of the Martin equation:

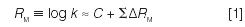
k is the retention factor for a substituted molecule (for example, m-ethyltoluene), C is the value of log k for the unsubstituted molecule (for example, benzene), and ΔRM is a constant for a given substitutent group (for example, methyl or ethyl). Later, we were asked to develop an assay of gasoline, which resulted in the development of "two-stage GC" (2) — an early form of column switching. By now, the power of chromatography, its connection to physical-organic chemistry, and the practical importance of method development were apparent.
Adsorption Chromatography and Petroleum Analysis (1957–1971)
In 1957, I moved to the Union Oil Co. of California, where my starting assignment was the analysis of high-boiling petroleum samples such as lubricating oil. Pioneering work of this kind had been carried out in O'Neil's group, where the sample was initially separated by adsorption chromatography into saturated hydrocarbons, aromatic hydrocarbons, and nonhydrocarbons. This was followed by the MS analysis of the hydrocarbon fractions. My experience at Shell suggested that previous such separations were empirically derived and poorly understood — so they likely could be improved. George Lake, my new supervisor at Union Oil, agreed and gave me a month to study the problem. The month grew into a separate 14-year research program and a 1968 book (Principles of Adsorption Chromatography [3]).
A simple equation summarized many of our findings,

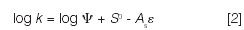
Here, k is a solute retention factor, Ψ is the phase ratio, As is the relative area of the solute molecule, and ε measures mobile-phase polarity or "solvent strength." S0 is the adsorption energy of the solute, values of which can be estimated as a function of solute molecular structure via equation 1 — in turn enabling predictions of k for different compounds and separation conditions.
My 1968 book would not be needed in today's HPLC laboratory, as information of present interest has since migrated into more recent books and publications. But it did describe a previously unappreciated phenomenon: adsorbate localization. Localization, the noncovalent attachment of polar molecules to specific adsorbent sites, was further studied over the next four decades (4) and found to largely determine solvent selectivity for adsorption chromatography (5) — an important contribution to method development for separations using silica or other adsorbents.
Petroleum consists mainly of hydrocarbons (alkanes, cycloalkanes, alkylbenzenes, and so on) plus lesser concentrations of related sulphur compounds (alkyl sulphides and thiophenes), and still smaller amounts of nitrogen- and oxygen-containing ("hetero") compounds. Our work on adsorption chromatography quickly led to improved compound-type separations of hydrocarbons plus sulphur compounds, for their subsequent MS analysis, as at Shell-Houston. This was accompanied by the development of complementary procedures for assaying certain other compound types in petroleum by so-called "linear elution adsorption chromatography" (6).
By 1966 we had completed work on the total analysis of hydrocarbons and sulphur compounds in any petroleum distillate, but our knowledge of the hetero compounds in such samples was incomplete. These oxygen- and nitrogen-containing molecules include several different compound types (such as, substituted pyridines, indoles, and furans), many of which were yet to be identified. Consequently, a complete analysis of these petroleum hetero compounds represented a major challenge. Our planned approach was the separation of different hetero-compound types from each other and from interfering hydrocarbons and sulphur compounds, followed by the spectral analysis of individual hetero fractions.
A multistep separation (7) made sequential use of three different adsorbents (alumina, silica, and charcoal) plus cation and anion exchange to provide about 50 hetero fractions. We could predict which compounds might be present in each fraction (equation 2), and this information with corresponding MS, UV, and infrared (IR) data resulted in the first comprehensive analysis of the N- and O-containing compounds in a petroleum distillate (8,9).
So, here we have an approach to pre-HPLC method development that relied on a quantitative knowledge of separation as a function of conditions for different possible compounds present in petroleum. This anticipated "efficient" method development, but is not applicable to HPLC, which deals with the more challenging separation of individual compounds, rather than compound types.
The Early Days of HPLC (1964–1971)
The arrival of HPLC in the mid-1960s has been described (for example, see reference 10). In 1966, I began packing columns with alumina for use with a crude, homemade HPLC system (11). An example of its subsequent use in our laboratory is shown in Figure 2 for the analysis of hydrogenated quinoline samples. An initial gradient separation [Figure 2(a)] revealed major peaks 1–5 that were eluted between 10 and 25 min. This suggested the routine isocratic separation of Figure 2(b), where an additional peak (peak 3a) is seen.

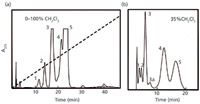
Figure 2: Analysis of hydrogenated quinoline by adsorption chromatography with alumina. (a) gradient elution (0â100% methylene chloride/n-pentane in 50 min); (b) isocratic separation (35% methylene chloride/n-pentane). Adapted from reference 12.
By 1970, the potential of HPLC had become obvious, but clearly its application within the petroleum industry would be limited. This realization — combined with the completion of our original petroleu-manalysis project — led me in 1971 to join the Technicon Corporation (Tarrytown, New York). At that time Technicon was the largest manufacturer of automated-analysis equipment for use in clinical laboratories.
It's worth recalling what HPLC looked like in 1971, as summarized in an early HPLC book (13). Commercial equipment and columns were available, and some of what had been learned about GC was being applied to LC. For example, the quality of a separation could be described by the resolution Rs of the two, poorest separated peaks in a chromatogram; Rs can be related (indirectly) to experimental conditions via retention factors k, separation factors α (selectivity), and the column plate number N (14):

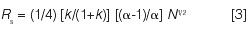
In the beginning, HPLC method development emphasized the improvement of resolution by predictable changes in conditions that affect values of k and N. In the case of N this might involve varying column length or flow rate (for example, see reference 15). The selection of preferred values of k (such as, 1 ≤ k ≤ 10) could be achieved by changing the proportions of a binary-solvent mobile phase; for reversed-phase chromatography an increase of organic solvent in the mobile phase by 10%v provides a predictable, 2–3-fold decrease in k (16).
The use of reversed-phase chromatography and gradient elution was quite limited in 1971; most HPLC separations were carried out isocratically by partition, adsorption, or ion exchange. Little was known of how best to change selectivity α, which is the most important parameter for controlling resolution. As in GC, random changes in the column (for example, stationary phase) were commonly tried at first. HPLC method development by "enlightened" trial-and-error was not often practiced, and a basis for "efficient" method development did not exist.
My Later Career (1971–2011)
From 1972 to 1977, I was VP of Research and later Clinical Chemistry at Technicon, with HPLC being downgraded to a "hobby interest" — however, the hobby was pursued with vigor. In 1971, Jack Kirkland and I presented the first ACS short course on HPLC ("Introduction to Modern Liquid Chromatography"), a two-day tutorial that continued during the next three decades with the later participation of John Dolan and Joe Glajch as co-instructors. Three editions of a book with the same title resulted (1974, 1979, and 2010 [17]), as well as two editions of a related book on method development (1987 and 1997 [18]). Both the course and the books proved popular, with > 6000 total attendees and 50000 books sold. An Introduction to Separation Science (Wiley) by Karger, Snyder and Horváth also appeared in 1973.
In 1977, I reduced my management responsibilities at Technicon and returned to hands-on R&D. Our immediate goal was an integrated HPLC system for the automated analysis of pharmaceutical drugs in serum — including sample preparation. The final equipment could be regarded as a technical success (19), but a market failure; automated immunoassay proved better suited for routine use. However, we maintained an active HPLC research program from 1977 until I left Technicon in 1982. Our group included John Dolan, Sjoerd van der Waal, Steve Bannister, and Russell Gant, aided by collaborations with Nobuo Tanaka and others from Barry Karger's group at Northeastern University.
Lloyd R. Snyder, Inc., was founded in 1982 as a one-man company based on HPLC. For the next seven years I also served as a consultant for DuPont Instruments in Wilmington, Delaware, which included supervising HPLC projects of my choice for four PhD students from nearby universities: Marilyn Stadalius (Delaware), Mary Ann Quarry and Julie Eble (Villanova), and Barbara Ghrist (Pennsylvania). In 1984, Lloyd R. Snyder, Inc., morphed into LC Resources with John Dolan as cofounder and headquarters in California; Tom Jupille joined us in 1987 as a third partner. Method development formed a significant part of our business from the beginning, with LC Resources growing to 30 employees by 2002. I also served as an editor of the Journal of Chromatography A from 1987 to 2000; Figure 3 shows several colleagues at my 2001 retirement party.


Figure 3: Participants at a symposium organized by the Journal of Chromatography A at Ellecom, The Netherlands (June 13â15, 2001), on the occasion of my retirement from the journal (20). Top row: P. Schoenmakers, J. Dolan, P. Carr, L. Snyder, J. Kirkland, I. Molnar, K. Bij, J. Dorsey, R. Giese, R. Marx, S. Terabe, N. Tanaka; middle row: E. Heftmann, D. McCalley, G. Vigh, R. Kaliszan, Cs. Horváth, S. Poole, S. Rutan, E. Soczewinski; bottom row: P. Jandera, C. Poole, H. Poppe, V. Davankov.
In 2002, LC Resources was split and sold to Rheodyne and Bioanalytical Systems. A few years later, the company was resurrected with the original three-man staff and an emphasis on short courses and collaborative research. From 1982 to present day, John, Tom and I taught HPLC to more than 10,000 individuals (apart from the ACS course), developed and marketed DryLab software (see below), and managed a method-development contract laboratory.
"Efficient" Method Development
By 1977, reversed-phase chromatography had become the primary HPLC procedure, and the use of gradient elution was becoming more common. However, further improvements in method development would require a better understanding of k, α, and N (equation 3) as a function of separation conditions, for both isocratic and gradient elution. "Efficient" method development (EMD) would not be possible until these relationships were made quantitative, so as to allow predictions (and optimization) of separation by a computer. A systematic approach to method development would also be needed.
Over time, a better understanding developed of which separation conditions have the greatest impact on selectivity (values of α). The importance and use of mobile-phase pH was well understood (for example, see reference 21), but only ionizable compounds are affected by pH. Less was known about other conditions that determine selectivity. EMD requires a choice of which separation conditions are best varied for an improvement in resolution. This choice must then be narrowed to specific solvents or columns, followed by preliminary separations. Finally, optimal conditions can be selected by some combination of further separations, experience, and computer simulation.
Work that eventually enabled EMD will be discussed next: advances in our understanding of selectivity as a function of separation conditions; a practical model of gradient elution; quantitative predictions of separation as a function of either isocratic or gradient conditions (computer simulation); and a method-development strategy. While many workers have contributed to our present knowledge, the following summary emphasizes those projects in which my group was involved.
Solvent Selectivity: The solvent-selectivity-triangle was introduced in 1973 (Figure 4), based on GC data by Rohrschneider for six test compounds and 80 solvents. This allowed the characterization of each solvent in terms of three possible interactions with the sample: hydrogen bonding (donor or acceptor) and "polar" (22).

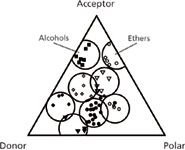
Figure 4: The solvent-selectivity triangle. Adapted from reference 22.
Three generalizations follow from Figure 4. First, as expected, solvents of similar functionality, for example, alcohols or ethers, fall within the same part of the triangle (grouped within circles). Second, a change in selectivity requires the use of a solvent from a different part of the triangle. Finally, it should be possible with mixtures of just three, well chosen solvents to approximately match the selectivity of any other solvent, for example, a reduction in the number of solvent choices from the original 80 to just three. The latter observation contributed in 1980 to the widely used four-solvent mapping system of Glajch and Kirkland for reversed-phase chromatography (23). Seven experiments allowed resolution to be predicted as a function of mobile-phase composition (mixtures with water of methanol, acetonitrile, and tetrahydrofuran), followed by the computer-selection of a "best" mobile phase. Based on this approach, the DuPont Sentinel system for "automatic" method development was introduced a few years later. Sentinel represented a big step toward "efficient" method development.
While the solvent triangle had its virtues, its derivation from data for pure solvents made it at best a qualitative model for the aqueous mobile phases of reversed-phase chromatography — as noted over the years (24). Indeed its ininitial application was intended for nonaqueous liquid–liquid chromatography, for which it was better suited. Other studies at about the same time also contributed to the selection of preferred solvents for controlling selectivity in reversed-phase chromatography (25,26). Today, methanol or acetonitrile is usually a first choice, with solvent selectivity optimized by varying their proportions in the mobile phase.
Solvent-Strength and Temperature Selectivity: Before 1985 it was widely assumed that controlling selectivity required changes in either the organic solvent, mobile-phase pH, or the column. That is, changes in either solvent strength (that is, the organic-solvent concentration, %B) or temperature were regarded as ineffective. In the mid-1980s, however, two different studies (27,28) showed that a change in isocratic %B or (equivalently) gradient steepness could in some cases provide marked changes in selectivity and successful separations. While isolated examples of temperature selectivity were known from the 1950s, its potential usefulness in reversed-phase chromatography method development was widely ignored. Some key experiments suggested by Bill Hancock in 1993 demonstrated that a change in temperature could be of great value for the separation of peptides (29). A few years later, we undertook a comprehensive study of the contribution of both %B and temperature to reversed-phase selectivity for various nonpeptide samples (30). Simultaneous changes in these two conditions were found to provide a convenient and generally effective control over selectivity (31), and for many laboratories this is now a preferred first step in method development. See the example of Figure 5 in a following section on computer simulation.


Figure 5: Computer simulation (DryLab) for the separation of a peptide mixture.
Column Selectivity: While a change of stationary phase can be an effective way of varying selectivity, at the present time there are > 1000 different reversed-phase columns from which to choose. A number of procedures for characterizing column selectivity were reported prior to 2000 (for example, see references 32–34), but with certain limitations (35). In 1998, John Dolan and I organized an exploratory meeting with Jack Kirkland, Pete Carr, John Dorsey, and Uwe Neue, from which a column-selectivity research plan resulted. This was followed by a 14-year program which later included the participation of Jon Gilroy and Chris Heard of Bioanalytical Systems, Dan Marchand at the University of Wisconsin–River Falls, Uwe Neue (Waters Inc., deceased), and David McCalley of the University of the West of England.
The latter work led to the development and application of the hydrophobic-subtraction (H-S) model of reversed-phase column selectivity, summarized recently in (35). By means of the H-S model, it is now possible to compare more than 500 reversed-phase columns in terms of selectivity, and to reliably choose columns whose selectivity is either similar or different using open source software (36). For routine assays, similar columns are needed as backup, while different columns can be used for orthogonal separation. Both kinds of columns are recommended for assays that fulfill "quality-by-design" (Loren Wrisley, cited in reference 37). The fundamental nature of the H-S model also supports several other uses related to method development — for example, problems with peak tailing and stationary phase degradation (35) — as well as provides a quantitative basis for understanding and using column selectivity.
Ion-Pairing: Ion-pair chromatography, as pioneered by John Knox and Goren Schill, provides still another opportunity to affect selectivity (see Chapter 7 of reference 17). This option is becoming less popular, however, because of the relative inconvenience of ion-pair chromatography (especially with MS detection) and the increasing replacement of ionpair chromatography by hydrophilic interaction chromatography (HILIC) (17).
Gradient Elution: Since the 1960s, chromatographers have been generally familiar with how isocratic separation is affected by changes in chromatographic conditions. A similar awareness for gradient elution emerged much later. Despite the derivation of the fundamental retention equation of gradient elution in 1955 by Drake, practical applications of gradient elution theory would remain dormant for the next 25 years. Our interest in gradient elution began in connection with adsorption chromatography. Starting with the Drake equation, an important concept became apparent by 1964: so-called linear-solvent-strength (LSS) gradients (38) that provide comparable peak spacing at both the start and finish of the separation.
In 1969, Dennis Saunders and I used computer modeling to arrive at a critical feature of LSS separation (39): as gradient steepness b is changed, resolution Rs is proportional to (1/b)/[(1/b)+1]. This is of the same form as a change in isocratic resolution with k (that is, k/[k+1]; see equation 3) and suggests that gradient steepness b is equivalent to 1/k in isocratic elution. A decade later, the LSS model was brought to initial completion with John Dolan and Russell Gant (40,41). Previous workers (for example, see reference 16) found that retention k in reversed-phase chromatography can be represented by

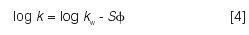
where kw is the value of k for φ = 0, S is a solute property that varies with molecular weight, and φ is the volume-fraction of mobile-phase organic solvent. This means that a linear reversed-phase gradient corresponds to LSS separation.
Some other practical conclusions followed for linear gradients. First, "average" values of k in gradient elution (k*) can be calculated from the gradient time tG, flow rate F, column dead-volume Vm, and Δφ (change in φ during the gradient):

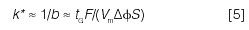
Very similar separations result for both isocratic and gradient elution when k ≈ k* for two adjacent peaks in the chromatogram. Consequently, our more highly developed understanding of isocratic selectivity has become equally applicable to gradient elution. Second, if the conditions of equation 5 are varied, but k* is held constant, separation selectivity will remain the same (important for method development). Finally, the LSS model proved critical for an understanding of the gradient separation of large biomolecules (for example, peptides and proteins) (42), as well as preparative separations by gradient elution (43,44). The development of the LSS model has continued since 1980 (44), with further contributions by Peter Schoenmakers, Hans Poppe, and especially Pavel Jandera.
Computer Simulation: By the early 1980s, various empirical or theoretical relationships (for example, equation 4) were known that described both isocratic and gradient retention as a function of separation conditions. This meant that a computer could be used to accurately predict (that is, simulate) changes in resolution when conditions are varied (45), in turn providing better separations with less work. In 1978, Laub and Purnell (46) described the related use of a "resolution map" [cf. Figure 5(a)] for ion-exchange HPLC as a function of temperature. A few years later (23), Glajch and Kirkland described the optimization of mobile phase composition as described above. At the same time, Deming and coworkers reported a similar approach for optimizing mobile-phase pH and the concentration of an ion-pair reagent (47). These various procedures presented plots of α vs. separation conditions, whereas maps of Rs are more useful; the latter resolution maps require predictions of N as a function of conditions.
By 1986, John Dolan and I had developed software (DryLab) for the accurate prediction of separation for any sample as a function of either isocratic %B or gradient time tG. Predictions of N as a function of experimental conditions were made possible by others' earlier studies plus refinements described in (48). As a result, DryLab could predict the result of changes in %B, tG, column dimensions, particle size, and flow rate (important variables in method development). Just two initial experimental separations were required, and in each case a chromatogram could be displayed for a change in any of these conditions (49). The software was later expanded to allow simultaneous changes in any two conditions (31,50), for example, varying temperature and either %B or gradient time, as in the example of Figure 5.
Four experimental separations at two different temperatures and gradient times are required initially. In Figure 5(a), a resulting resolution map is shown for a mixture of 19 peptides (digest of the protein rhGH [29]). Regions of higher resolution (see key on left) and shorter gradient time are preferred, so the cross-hairs are set as shown: 43 °C and tG = 64 min. A corresponding chromatogram is automatically displayed [Figure 5(b)], with a run time of 40 min and a resolution Rs = 3.5. Because there is an excess of resolution, run time can be reduced. The software is next used to explore the use of a shorter column or higher flow rate, each of which will decrease run time (at the expense of resolution). Figure 5(c) shows a separation for a reduction in column length from 150 to 50 mm, an increase in flow rate from 2 to 4 mL/min, and a computer-selected gradient time of tG = 11 min (the latter maintains k* and selectivity constant; equation 5). Baseline resolution (Rs = 1.5) is accompanied by a sixfold shorter run time.
Later versions of DryLab have expanded these various capabilities (for example, see Chapter 10 of reference 17). In 2002, DryLab was sold by LC Resources to Rheodyne and in 2006 became the property of the Molnar Institute (Berlin). Recent improvements in DryLab allow the simultaneous optimization of three different separation conditions (for example, gradient time, temperature, and pH), using 12 experiments (51).
Method Development Strategy: A method development strategy depends on the nature of the sample, the chromatographer's experience (especially with related samples), and the goals of separation. Any method development plan must therefore be quite flexible. As summarized in (17), EMD best begins with the selection of suitable gradient conditions: for example, a 0–100% gradient with a low-pH mobile phase and acetonitrile as organic solvent, a temperature of 30–40 °C, and a C18 column packed with ~3-µm particles.
Selectivity is next optimized, while maintaining reasonable retention factors (for example, 1 = k* = 10; equation 5). Four initial experiments that vary temperature and gradient steepness (as in Figure 5) can be carried out. For more demanding separations, additional experiments are added which explore other conditions for changing selectivity (pH, solvent or column type). Once "promising" selectivity has been achieved, the plate number N can be optimized (by change in flow rate, column length, or particle size) to provide the best trade-off between resolution and run time as in the example of Figure 5(c). At each step, computer simulation is used, which also allows predictions of isocratic separation from gradient data. (A similar approach can be used for normal-phase and ion-exchange chromatography [17], also supported by DryLab.)
What Next?
"Efficient" method development is now possible in any laboratory (17), based on the combined use of computer simulation, our present understanding of HPLC separation, and a method development strategy. The next logical step is "automatic" method development, using an HPLC system under the total control of a computer. The computer first requests information about the sample and separation goals, then selects conditions for various experiments. Results from initial experiments are interpreted, further experiments are designed and carried out as needed, and "best" conditions are inferred (all by computer). A final experimental separation then serves to confirm the recommended conditions. The DuPont Sentinel system (see above) was a good start in this direction, but was limited to changes in solvent selectivity.
As every practical worker knows, there is more to method development than simply choosing separation conditions, for example, sample preparation, choice of detector, method robustness, and so on (17,18). Some more ambitious attempts in this direction ("expert systems") were described in the late 1980s (52), but never achieved acceptance. Since 2000 several systems for "automated" method development have been marketed, but fall short of "efficient" method development. The skill and experience of the individual chromatographer have not yet become redundant.
Model building and its chromatographic applications pretty much sum up my career, as illustrated by the above projects. Many people were involved in this work, notably John Dolan, Pete Carr, and Jack Kirkland. In Pete Carr's recent LCGC biography (53), he described two kinds of chromatographers: kineticists and thermodynamicists. Like Pete, I am clearly a "thermodynamicist," while Jack is more of a "kineticist," and John defies classification . . . viva la difference! Last and not least, little of my work would have happened without favorable circumstances and relationships, and the support of Barbara — my wife of 60 years. From start to finish, fortune smiled on these endeavors.
Lloyd R. Snyder received his B.S. and PhD degrees in chemistry from the University of California at Berkeley in 1952 and 1954. Since 1954 he has worked on the theory and practice of chromatography, as well as HPLC instruction via short courses and books. His work has received national and international recognition, including American Chemical Society awards in Petroleum Chemistry (1970) and Chromatography (1984), the Pittsburgh Conference Analytical Chemistry (1984), the A.J.P. Martin award of the Chromatographic Society (1989) and the Waksmundzki Medal of the Polish Analyical Society (2005).
References
(1) D.S. Noyce and L.R. Snyder, J. Amer. Chem. Soc. 80, 4033, 4324; 81, 620 (1958–1959).
(2) M.C. Simmons and L.R. Snyder, Anal. Chem. 30, 32 (1958).
(3) L.R. Snyder, Principles of Adsorption Chromatography. The Separation of Nonionic Organic Compounds, (Marcel Dekker, New York, 1968).
(4) L.R. Snyder, J. Planar Chromatogr. 25, in press (2012).
(5) L.R. Snyder, J. Planar Chromatogr. 21, 315 (2008).
(6) L.R. Snyder and D.L. Saunders, in Petroleum Analysis, K.H. Altgelt and T.H. Gouw, Eds. (Dekker, New York, 1979), Chap. 10.
(7) L.R. Snyder and B.E. Buell, Anal. Chem. 40, 1295 (1968).
(8) L.R. Snyder, B.E. Buell and H.E. Howard, Anal. Chem. 40, 1303 (1968).
(9) L.R. Snyder, Accts. Chem. Res. 3, 290 (1970).
(10) L.R. Snyder and J.W. Dolan, Handbook of Separation Science: Liquid Chromatography, (Elsevier, Amsterdam, 2012), Chap. 1.
(11) L.R. Snyder, Anal. Chem. 39, 698, 705 (1967).
(12) L.R. Snyder, J. Chromatogr. Sci. 7, 595 (1969).
(13) Modern Practice of Liquid Chromatography, J.J. Kirkland, Ed. (Wiley-Interscience, New York, 1971).
(14) J.H. Purnell, J. Chem. Soc. 1268 (1960).
(15) L.R. Snyder, J. Chromatogr. Sci. 10, 200, 369 (1972).
(16) J.A. Schmidt, R.A. Henry, R.C. Williams and J.F. Dieckmann, J. Chromatogr. Sci. 9, 645 (1971).
(17) L.R. Snyder, J.J. Kirkland and J.W. Dolan, Introduction to Modern Liquid Chromatography, 3rd Ed. (WileyInterscience, New York, 2010).
(18) L.R. Snyder, J.J. Kirkland and J.L. Glajch, Practical HPLC Method Development, 2nd Ed. (Wiley Interscience, New York, 1997).
(19) Sj. van der Wal, S.J. Bannister, J.W. Dolan and L.R. Snyder, Clin. Chem. 26, 871 and 27, 849 (1980–1981).
(20) "Application of Theory to the Practice and Understanding of Chromatography" J.G. Dorsey, Ed., J. Chromatogr. A 965 (2002).
(21) J.A. Lewis, D.C. Lommen, W.D. Raddatz, J.W. Dolan, L.R. Snyder and I. Molnar, J. Chromatogr. 592, 183 (1992).
(22) L.R. Snyder, J. Chromatogr. 92, 223 (1974).
(23) J.L. Glajch, J.J. Kirkland, K.M. Squire and J.M. Minor, J. Chromatogr. 199, 57 (1980).
(24) A.R. Johnson and M.F. Vitha, J. Chromatogr. A 1218, 556 (2011).
(25) S.R. Bakalyar, R. McIlwrick and E. Roggendorf, J. Chromatogr. 142, 353 (1977).
(26) N. Tanaka, H. Goodell and B.L. Karger, J. Chromatogr. 158, 233 (1978).
(27) M.A. Quarry, R.L. Grob, L.R. Snyder, J.W. Dolan and M. Rigney, J. Chromatogr. 384, 163 (1987).
(28) J.L. Glajch, M.A. Quarry, J.F. Vasta and L.R. Snyder, Anal. Chem. 58, 280 (1986).
(29) W. Hancock, R.C. Chloupek, J.J. Kirkland and L.R. Snyder, J. Chromatogr. A 686, 31, 45 (1994).
(30) P.L. Zhu, L.R. Snyder, J.W. Dolan et al, J. Chromatogr. A 756, 21, 51, 63 (1996).
(31) J.W. Dolan, L.R. Snyder, N.M. Djordjevic, D.W. Hill, D.L. Saunders, L. Van Heukelem and T.J. Waeghe, J. Chromatogr. A 803, 1 (1998).
(32) P.C. Sadek, P.W. Carr, R.M. Doherty, M.J. Kamlet, R.W. Taft and M.H. Abraham, Anal. Chem. 57, 2971 (1985).
(33) K. Kimata, K. Iwaguchi, S. Onishi, K. Jinno, R. Eksteen, K. Hosoya, M. Araki and N. Tanaka, J. Chromatogr. Sci. 27, 721 (1989).
(34) E. Cruz, M.R. Euerby, C.M. Johnson and C.A. Hackett, Chromatographia 44, 151 (1997).
(35) L.R. Snyder, J.W. Dolan, D.H. Marchand and P.W. Carr, Adv. Chromatogr. 50, Chap. 7 (2012).
(36) United States Pharmacopeia website (http://www.USP.org/USPNF/columns.html), PQRI version.
(37) L.R. Snyder, P.W. Carr, J.W. Dolan and R.E. Majors, LCGC North America 28, 418 (2010).
(38) L.R. Snyder, J. Chromatogr. 13, 415 (1964).
(39) L.R. Snyder and D.L. Saunders, J. Chromatogr. Sci. 7, 195 (1969).
(40) L.R. Snyder, J.W. Dolan and J.R. Gant, J. Chromatogr. 165, 3 (1979).
(41) L.R. Snyder, in High-Performance Liquid Chromatography. Advances and Perspectives, Vol. 1, Cs. Horváth, Ed. (Academic Press, New York, 1980), Chap. 4.
(42) L.R. Snyder and M.A. Stadalius, in High-Performance Liquid Chromatography. Advances and Perspectives, Vol. 4, Cs. Horváth, Ed. (Academic Press, New York, 1986), p. 195.
(43) J.E. Eble, R.L. Grob, P.E. Antle and L.R. Snyder, J. Chromatogr. 405, 51 (1987).
(44) L.R. Snyder and J.W. Dolan, High-Performance Gradient Elution, (Wiley-Interscience, Hoboken, New Jersey, 2007).
(45) J.R. Gant, J.W. Dolan and L.R. Snyder, J. Chromatogr. 185, 153 (1979).
(46) R.J. Laub and J.H. Purnell, J. Chromatogr. 161, 49 (1978).
(47) B. Sachok, R.C. Kong and S.N. Deming, J. Chromatogr. 199, 317 (1980).
(48) R.W. Stout, J.J. DeStefano and L.R. Snyder, J. Chromatogr. 282, 263 (1983).
(59) L.R. Snyder and J.W. Dolan, LCGC North America 5, 970 (1987).
(50) P. Haber, T. Baczek , R. Kaliszan, L.R. Snyder, J.W. Dolan and C.T. Wehr., J. Chromatogr. Sci. 38, 386 (2000).
(51) I. Molnár, H.-J. Rieger and K.E. Monks, J. of Chromatogr. A 1217, 3193 (2010).
(52) "Computer-assisted Chromatographic Method Development" J.L. Glajch and L.R. Snyder, Eds., J. Chromatogr. 485, (1989).
(53) P.W. Carr, LCGC North America 28, 610 (2010).















Determining the Serum Proteomic Profile in Migraine Patients with LC–MS
April 17th 2025Researchers used liquid chromatography–mass spectrometry (LC–MS) in their proteomic analysis to compare the serum proteome of migraine patients with healthy controls and to identify differentially expressed proteins as potential migraine biomarkers.


