Comprehensive Two-Dimensional Liquid Chromatography Coupled to Triple Quadrupole Mass Spectrometry: Application to a Challenging Food Case Study
Special Issues
The complexity of many food samples places a great demand in terms of both separation capabilities and specificity of detection. In this article, a novel system for fully automated comprehensive two-dimensional liquid chromatography (LC?LC) is discussed.
The complexity of many food samples places a great demand in terms of both separation capabilities and specificity of detection. In this article, a novel system for fully automated comprehensive two-dimensional liquid chromatography (LC×LC) is discussed. The on-line coupling of the two separation dimensions was achieved using two six-port, two-position switching valves. High orthogonality was achieved by using a micro-bore cyano column for the first dimension separation, interfaced to a secondary C18 column packed with fused-core particles. The hyphenation to a triple quadrupole mass spectrometer generates a powerful analytical system, capable of extremely high resolving power, as well as targeted and untargeted analysis. The so-called multiple reaction monitoring (MRM) mode in fact enhanced selectivity, reducing sample consumption and the need for tedious clean-up procedures, specifically for beta-carotene quantification in a red pepper extract.
The high complexity of many food samples places a great demand in terms of both separation capabilities and specificity of detection. As far as separation is concerned, the implementation of multidimensional liquid chromatography (LC) techniques has provided enhanced resolving power for highly complex samples, especially in the "comprehensive" mode (LC×LC), in which the whole effluent from the first chromatographic dimension (1D) is transferred to a second chromatographic dimension (2D). As far as operation mode is concerned, "continuous on-line" techniques bring in additional advantages, including no need for flow interruption, no increase in overall analysis time, and full automation of two-dimensional LC; for example, involving off-line transfer between the two dimensions, or the "stop-flow" techniques (1–5).
The coupling of LC×LC separation to mass spectrometry (MS) detection generates the most powerful analytical tool for non-volatile analytes. Such a combination offers a number of analytical advantages, and may help to overcome some limitations of the two techniques, when considered individually. With respect to conventional LC–MS, the combination of two LC separations enhances physical separation of the components, reducing undesirable matrix effects arising from co-elutions. Maximizing the resolution is in fact beneficial for subsequent MS detection, in terms of sensitivity and dynamic range, since it alleviates ion suppression effects resulting from insufficient separation, which may cause high abundant species to obscure the detection of less abundant ones. Unlike the UV detector, MS systems can also be used with non-absorbing analytes, and can be operated in the full scan mode (TIC) or, more specifically, in tandem MS (MS–MS) experiments or in the selected ion monitoring (SIM) mode. Constant neutral loss or precursor ion scanning techniques help to distinguish the ions of interest from unspecific matrix components by monitoring only those m/z values that originate from a characteristic fragmentation pattern. The so-called multiple reaction monitoring (MRM) mode enhances selectivity and lowers detection limits, therefore reducing sample consumption; in addition, the MRM approach can also decrease analysis times by reducing the need for clean-up procedures, which are often mandatory, prior to the analysis of complex samples, such as many foodstuffs (6).
Among these complex samples, carotenoids represent a challenging analysis task for a number of reasons. These include high variability in the chemical structures, isomerization, poor stability, and the lack of commercially available standards for reliable identification and quantification in real samples. Commonly found in plants, algae, fungi, and bacteria, carotenoids consist of a C40-tetraterpenoid structure with a symmetrical skeleton (7), and are usually divided into two groups: Hydrocarbon carotenoids, generally known as carotenes (such as β-carotene, lycopene), and oxygenated carotenoids, known as xanthophylls (for example, lutein, β-cryptoxanthin) (8). These compounds can be found in nature in their free form, or in a more stable fatty acid esterified form. The study of esterified carotenoids in natural sources is rather limited, especially because of the high degree of complexity; a saponification step is instead often used prior to LC analysis. However, such a strategy does present some drawbacks as during the saponification procedure, strong conditions are used and, as a consequence, carotenoid loss, as well as isomerization, can occur (9).
As far as separation is concerned, reversed-phase LC with both C18 and C30 stationary phases has been extensively used to achieve the separation of molecules differing in hydrophobicity within a given structural class (10). On the other hand, normal-phase LC on silica-based columns is largely used for carotenoid class separation, according to different polarity (with retention increasing from hydrocarbons to xanthophylls). A major limitation of this approach consists in diminished resolving power when separating carotenoid classes lying at the two extremes of polarity scale (hydrocarbons and xanthophylls [11]) from the rest of the matrix.
Furthermore, the true potential of coupling different C18 columns (12) and two-dimensional LC approaches (13,14) has been investigated to increase the separation power and thus resolution and efficiency for the analysis of carotenoids in extremely complex natural samples.
Identification of carotenoids in foodstuffs is generally accomplished by the complementary information provided by LC retention times, photodiode array (PDA), and MS data. Although commonly used for carotenoid identification, PDA detection nevertheless fails in the case of analytes which exhibit similar — or even identical — spectra. On the other hand, MS was excellent for the analysis of these substances, allowing structure elucidation on the basis of both molecular mass and fragmentation pattern. Several ionization methods have been reported for MS analysis of carotenoids, including electron impact (EI), fast atom bombardment (FAB), matrix-assisted laser desorption–ionization (MALDI), electrospray ionization (ESI), atmospheric-pressure chemical ionization (APCI) and, more recently, atmospheric-pressure photoionization (APPI) and atmospheric pressure solids analysis probe (ASAP) (15). Sometimes, LC–MS–MS or MSn can be advantageously applied to carotenoid analysis through the use of specific transitions and daughter ions for the identification of analytes via precursor ion selection, eventually allowing carotenoids of equal molecular masses but different fragmentation patterns to be discriminated between (16).
This article describes a novel LC×LC–PDA–MS–MS system capable of extremely high-resolving power, as well as targeted and untargeted analysis, simultaneously. The system was successfully used for the characterization of native carotenoids in red chili pepper, in addition to quantification of beta-carotene at sub-ppm level.
Experimental
Standards and Chemicals
Beta carotene reference material, as well as LC–MS grade n-hexane, butylacetate, acetone, acetonitrile, and isopropanol, were obtained from Sigma-Aldrich/Supelco. Methanol, ethyl acetate, petroleum ether, and tert-butyl methyl ether (all reagent-grade) were supplied by Sigma-Aldrich.
Sample and Sample Preparation
The red chili pepper sample (Capsicum annuum L.) was purchased in a local market. For the extraction of intact carotenoids (not saponified), 200 g of red chili pepper homogenate were treated with three consecutive 300 mL aliquots of a methanol/ethyl acetate/petroleum ether (1:1:1, v/v/v) mixture. The extracts were combined and filtered through a paper filter. They were then evaporated to dryness under vacuum (30 °C), and the dry residue dissolved in 3 mL of a methanol/tert-butyl methyl ether (1:1, v/v) mixture and filtered through a 0.45 µm Acrodisc nylon membrane (Pall Life Sciences).
LC×LC–MS–MS Analysis
LC×LC analyses were performed on a comprehensive two-dimensional LC×LC system (Shimadzu), consisting of a CBM-20A controller; four LC-30AD dual-plunger parallel-flow pumps; a DGU-20A5R degasser; a DGU-20A3R degasser; a CTO-20AC column oven; a SIL-30AC autosampler; and a SPD-M30A photo diode array detector (1.8-µL detector flow cell volume). The two dimensions were connected by two high speed/high pressure two-position, six-port switching valves with micro-electric actuator (model FCV-32 AH, 1.034 bar; Shimadzu), placed inside the column oven and equipped with two 20-µL (non-packed) stainless steel loops. Both dimensions and the switching valve were controlled by the Shimadzu Labsolution software (ver. 5.60 SP2). The LC×LC data were visualized and elaborated into two and three dimensions using Chromsquare ver. 2.0 software (Shimadzu Corporation). The LC×LC system was coupled to an LCMS-8030 triple quadrupole mass spectrometer through a DUIS source (Shimadzu). Figure 1 shows a schematic of the two-dimensional system employed in this work, with the switching valves equipped with the sample loops.


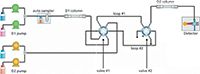
Figure 1: Schematic of the LCÃLCâPDAâMSâMS system, with high pressure two-position, six-port switching valves equipped with non-packed sample loops.
For the first dimension separation, a 250 × 1.0 mm, 5-µm dp Ascentis ES-Cyano column was used (Sigma-Aldrich/Supelco). Mobile phases: (A) n-hexane, (B) n-hexane/butylacetate/acetone (80/15/5, v/v/v). Gradient: 0–5 min, 0% B, 5–65 min, to 100% B. Mobile phase flow rate: 20 µL/min. Column oven: 30 °C. Injection volume: 2 µL.
For the second dimension separation, a 50 × 4.6 mm, 2.7-µm dp Ascentis Express C18 column was used (Sigma-Aldrich/Supelco). Mobile phases: (A) acetonitrile, (B) isopropanol. Gradient: 0.01 min, 0% B, 0.01–0.17 min, to 50% B, 0.17–0.27 min, 50% B, 0.27–0.54 min, to 80% B, 0.54–0.93 min, 80% B, 0.94 min, to 30% B. Mobile phase flow rate: 4 mL/min. Column oven: 30 °C. Modulation time of the switching valves: 60 s. PDA detection range: 250–550 nm; sampling rate: 12.5 Hz; time constant: 0.080 s. MS detection: DUIS positive mode; full-scan mass spectral range: 410–1200 m/z; event time: 0.1 s; nebulizing gas (N2) flow: 2.5 L/min; drying gas (N2) flow: 20 L/min; heat block temperature: 400 °C; desolvation line (DL) temperature 250 °C; interface voltage 4.5 kV. For the identification of selected compounds, product ion scan experiments were performed. For the quantitative determination of beta-carotene, MS parameters were optimized for MRM of the following transitions: m/z 536.40 to m/z 444.30 (quantifier ion) and m/z 536.40 to m/z 105.00 (qualifier ion).
From the LC system, 800 µL/min of the 2D flow were directed to the probe. For the splitting device, a stainless steel microvolume connector was used (1/16", 0.15-mm bore) from VICI (Valco Instruments Co. Inc.).
Results and Discussion
This research was performed to develop an on-line LC×LC–PDA–MS–MS system, based on triple quadrupole technology, capable of fully automated operation, and exploitable for the analysis of highly complex samples, both in the targeted and untargeted mode. This novel system was assessed for the analysis of the carotenoid content of a red chili pepper extract, and the quantitative determination of the main bioactive molecule, beta-carotene.
Such a sample was chosen because of its complexity, by far overwhelming the limited peak capacity afforded by any monodimensional LC technique. Multidimensional techniques are therefore deemed as necessary, not only from the separation standpoint, but also as far as MS detection is concerned. Step-by-step implementation of the system addressed the following issues: Choice of the stationary phases, column dimensions, flow rates, connections, and detection parameters.
In our LC×LC platform, normal-phase LC and reversed-phase LC were used in the first and the second dimension, respectively. Normal-phase LC × reversed-phase LC represents one of the most orthogonal approaches in two-dimensional LC, capable of delivering very high resolution power as a result of independent separation mechanisms operating on the individual stationary phases. However, this type of coupling is not easy to achieve because of the incompatibility of the solvents that are used in the two dimensions, and possible problems of peak focusing at the head of the secondary column (13,14). This issue was circumvented by the use of a micro-bore column in the first dimension. This served two purposes: Firstly, to reduce the mobile phase immiscibility and the occurrence of solvent strength mismatch, which would have affected system compatibility in such a combination. Non-polar hexane was in fact the 1D solvent, while aqueous acetonitrile and isopropanol mixtures were employed for the 2D analysis. In addition, the low flow rate employed also reduced band broadening and helped to achieve effective peak focusing at the head of the 2D column, thanks to the minimized mobile-phase strength, connected to the transfer from 1D to 2D. On the other hand, the separation in 2D must be fast and on-column focusing must be achieved in the on-line comprehensive multidimensional system. The fully automated LC×LC system was configured around two electronically activated two-position, six-ports switching valves for within-loop automated fraction collection and re-injection, equipped with two storage loops of identical volume (20 µL). The whole effluent from the 1D micro-bore column was transferred on-line to the second dimension column.
The first dimension effluent was fractionated every 60 s, which was the modulation time (cycle time) of the switching valves, which in turn was equal to the analysis time in 2D. The choice of the modulation time and the gradient profile was made to meet the following stringent requirements: Each fraction injected onto the second dimension column must be completely eluted before the following transfer occurred; the 2D analysis time had to be kept as short as possible, so as not to impair the separation achieved in the 1D (in this respect, a large number of cuts is highly beneficial). Moreover, re-conditioning of the stationary phase from each repetitive gradient should be fast enough to fit the limited time allowed.
In order to fulfil all these requirements, the second dimension column was operated at a very high flow rate (4 mL/min), with a gradient programme starting with 100% concentration of the weaker solvent, acetonitrile. A fast ramp up to 80% of the stronger solvent (isopropanol) was run to ensure the elution of all the components within a fast analysis time of 60 s, including a reconditioning time of 0.06 min. Repetitive injections of the sample under such conditions afforded perfectly super-imposable elution profiles (data not shown), demonstrating sufficient column re-equilibration time after the gradient. The shortest possible length of stainless steel tubing of 0.1 mm i.d. was used for extra-column connections and valves; as predicted, longer or higher internal diameter tubing negatively affected the overall performance of the system. This was in part as a result of additional dead volumes and the resulting peak broadening, but also down to significant dilution which jeopardized the MS response, with the overall dilution factor multiplicative of the dilution factors in each dimension. When designing an on-line LC×LC–MS technique, the detector response and the limited dynamic range of the instrument must also be considered. A fast acquisition rate was necessary, both for the PDA and the MS detector, because the second-dimension separation was very rapid, and peak widths only a few seconds or less apart. To adequately sample the 2D effluent, the mass analyzer should be capable of acquiring at least 6–10 data points for each peak. While traditional quadrupoles suffered from low resolution (typically 1 Da) and presented a mass range limited to approximately m/z 4000, the instrumentation used here allowed for high speed scanning (up to 15,000 amu/s) and ultrafast polarity switching for high-speed analyses, in both positive and negative ion mode.
Results obtained from the normal-phase LC × reversed-phase LC analysis of free carotenoids and carotenoid esters in the red chili pepper extract are shown in the contour plot of Figure 2. Chromatography on the cyano stationary phase allowed a good separation of the carotenoids in groups of different polarity in the first dimension, as can be seen from the ellipses in Figure 2, with retention times increasing in the order: Hydrocarbons < monool esters < diol diesters < diol monoketo diesters < diol diketo diesters < diol monoepoxide monoesters < free monools < diol monoketo monoesters < diol diketo monoesters < polyoxygenated free xanthophylls. On the other hand, the C18 column allowed the separation of carotenoids within each class, according to their increasing hydrophobicity and decreasing polarity (for components of the same class, the elution order increased with the number of carbon atoms of the fatty acid chain). By optimizing the elution parameters (as a compromise between the chromatographic separation, modulation time, maximum column operating temperature, and highest flow rate allowed by the system back pressure limit), a satisfactory separation of the sample carotenoids was obtained, with 10 different classes distributed along characteristic chemical patterns in the 2D retention plane.


Figure 2: Normal-phase LC Ã reversed-phase LC contour plot of free carotenoids and carotenoid esters in the red chilli pepper extract.
Identification of the separated compounds was achieved by both PDA and MS data. The possibility to operate in both positive ion and negative ion mode is a useful feature for the untargeted analysis of unknown samples, and in the special case of carotenoids it offered the double advantage of improved sensitivity and identification power. MS spectra obtained under negative ionization mode are in fact dominated by the presence of very intense pseudomolecolar ions [M].-, which make identification and quantitation of low-abundant components easier; on the other hand, abundant fragmentation is generally observed under positive ionization, especially for carotenoid esters, whose fragment ions can help in structure elucidation through a dedicated software or database.
The carotenoid content of this extracted red chili pepper, as determined by the method developed, was qualitatively very similar to that determined in our previous work (14), with only a few exceptions. The first regarded the absence of the chemical class of the diol monoepoxide monoesters; in contrast an additional carotenoid class was found in this cultivar — the diol diketo monoesters. Capsorubin monoester with myristic acid (C14:0), whose chemical structure is shown in Figure 3, is representative of this chemical class, which comprises very polar molecules and elutes almost at the end of the first dimension gradient, immediately before the xanthophylls (ellipse 9 in Figure 2). Its UV spectrum shows a single absorption maximum centred at 473 nm, while the protonated molecular ion ([M+H]+) at m/z 811.2 showed a weak signal in the MS spectrum as a result of intense fragmentation, depicting the expulsion of water and myristic acid moieties in the product ion scan experiment (18 and 228 Da). Occasionally, these losses were also observed in the full scan experiment as a result of in-source fragmentation. In all cases, the formation of dehydrated product ions seemed to be favored, confirming the presence of a non-esterified hydroxyl function. An intense signal was detected as well, at m/z 713.4 [M-18-79], which according to the literature, may derive from the loss of a water molecule and the in-chain elimination of a C6H7 fragment ion that occurs via a common seven-membered ring transition state (17).

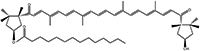
Figure 3: Chemical structure of capsorubin ester with myristic acid.
The main advantage of this developed system, with respect to the one previously used (14), lies in the possibility of carrying out quantitative analysis with very high selectivity and sensitivity, in the so-called MRM mode, for target molecules. Beta-carotene (m/z 536.40) transitions at m/z 444.30 (quantifier ion) and m/z 105.00 (qualifier ion) were selected for the construction of a calibration curve, in the 1 ppb to 10 ppm concentration range. Very good linearity was observed, with regression line equation: y = 410259x + 49414; linear correlation coefficient (R) = 0.998976. The amount of beta-carotene was determined afterwards in the real sample as equal to 1.22 ppm. Figure 4 shows an expansion of the 2D plot, with the beta-carotene blob and the MS spectrum with the two transitions.

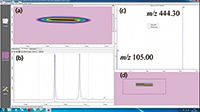
Figure 4: Enlargment of the 2D contour plot illustrated in Figure 2. (a): Beta-carotene at m/z 536.40. (b): Raw data of three consecutive modulation times (60 s each). (c): MS spectrum showing the transitions at m/z 444.30 (quantifier ion) and m/z 105.00 (qualifier ion). (d): Non interpolated 2D data.
Conclusions
In this article, an LC×LC system was coupled on-line to a triple quadrupole mass spectrometer, developing a novel analytical system capable of extremely high-resolution power, as well as targeted and untargeted analysis. For the separation, identification, and quantification of highly complex samples, unique features of this system consist of: High resolving power, fast analysis time, full automation, high sensitivity and selectivity, fragment information, and fast cycle time (including polarity switching).
Moreover, the 2D plots rendered by the front-end LC×LC separation may be of great help in the identification of unknowns, whenever standard components or reliable MS library spectra are not available. The separated molecules are in fact distributed along chemically-similar compound patterns, and this will provide useful information on their structures and properties, according to the separation mechanisms involved.
Paola Donato is an assistant professor of analytical chemistry at the University Campus Bio-Medico of Rome, Italy. Her research activity is mainly focused on the development of multidimensional liquid chromatography instrumentation coupled to mass spectrometry for the analysis of highly complex samples.
Francesco Cacciola is an assistant professor of food chemistry at the University of Messina, Italy. His research interests include the characterization of complex food samples by conventional and innovative LC and MS techniques.
Francesca Rigano, PhD, is a student in food chemistry and safety at the University of Messina, Italy. Her research is mainly focused on the development of nano- and comprehensive liquid chromatography instrumentation coupled to electron ionization and triple quadrupole mass spectrometry, respectively.
Daniele Giuffrida is an assistant professor of organic chemistry at the University of Messina, Italy. His research activity is mainly focused on the investigation of bioactive molecules in natural products and foodstuffs by means of advanced and multidimensional chromatographic techniques .
Paola Dugo is a full professor of food chemistry at the University of Messina, Italy. Her research interests include the development of comprehensive liquid chromatography techniques and high throughput separation methods for the study of food components with potential biological activity.
Luigi Mondello is a full professor of analytical chemistry at the University of Messina, Italy. His research interests include chromatographic techniques, hyphenation to mass spectrometry, and multidimensional chromatographic approaches for the study of complex real-world samples. Direct correspondence to: lmondello@unime.it
References
(1) P. Dugo, F. Cacciola, T. Kumm, G. Dugo, and L. Mondello, J. Chromatogr. A 1184, 353–368 (2008).
(2) G. Guiochon, M. Marchetti, K. Mriziq, and R.A. Shalliker, J. Chromatogr. A 1189, 109–168 (2009).
(3) I. François, K. Sandra, and P. Sandra, Anal. Chim. Acta 641, 14–31 (2009).
(4) F. Cacciola, P. Donato, M. Beccaria, P. Dugo, and L. Mondello, LCGC Eur. 5, 15–24 (2012).
(5) P.Q. Tranchida, P. Donato, F. Cacciola, M. Beccaria, P. Dugo, and L. Mondello, Trend Anal. Chem. 52, 186–205 (2013).
(6) P. Donato, F. Cacciola, P.Q. Tranchida, P. Dugo, and L. Mondello, Mass Spectrom. Rev. 31, 523–559 (2012).
(7) F. Delgado-Vargas, A.R. Jiménez, and O. Paredes-López, Crit. Rev. Food Sci. 40, 173–289 (2000).
(8) D. Rodriguez-Amaya and M. Kimura, Harvest Plus Handbook for Carotenoids Analysis (Harvest Plus, Washington, DC., USA, 2004).
(9) P. Dugo, V. Skerikova, T. Kumm, A. Trozzi, P. Jandera, and L. Mondello, Anal. Chem. 78, 7743–7750 (2006).
(10) S.M. Rivera and R. Canela-Garayoa, J. Chromatogr. A 1224, 1–10 (2012).
(11) G. Panfili, A. Fratianni, and M. Irano, J. Agric. Food Chem. 52, 6373–6377 (2004).
(12) M. Herrero, F. Cacciola. P. Donato, D. Giuffrida, G.mo Dugo, P. Dugo, and L. Mondello, J. Chromatogr. A 1188, 208–215 (2008).
(13) P. Dugo, D. Giuffrida, M. Herrero, P. Donato, and L. Mondello, J. Sep. Sci. 32 973-980 (2009).
(14) F. Cacciola, P. Donato, D. Giuffrida. G. Torre, P. Dugo, and L. Mondello, J. Chromatogr. A 1255, 244–251 (2012).
(15) S.M. Rivera and R. Canela-Garayoa, J. Chromatogr. A 1224, 1–10 (2012).
(16) G. Oms-Oliu, I. Odriozola-Serrano, and O. Martin-Belloso, Food Res. Int. 54, 1172–1183 (2009).
(17) G. Britton, S. Liaaen-Jensen, and H. Pfander, Carotenoids Volume 1B, Spectroscopy (Birkhauser Verlag Basel, Switzerland, 1995).














Analytical Challenges in Measuring Migration from Food Contact Materials
November 2nd 2015Food contact materials contain low molecular weight additives and processing aids which can migrate into foods leading to trace levels of contamination. Food safety is ensured through regulations, comprising compositional controls and migration limits, which present a significant analytical challenge to the food industry to ensure compliance and demonstrate due diligence. Of the various analytical approaches, LC-MS/MS has proved to be an essential tool in monitoring migration of target compounds into foods, and more sophisticated approaches such as LC-high resolution MS (Orbitrap) are being increasingly used for untargeted analysis to monitor non-intentionally added substances. This podcast will provide an overview to this area, illustrated with various applications showing current approaches being employed.
 Headquarters complex in Washington, DC. | Image Credit: © Tada Images - stock.adobe.com")




