Analytical Applications of Immobilized Enzyme Reactors (IMERs) Coupled to LC–MS/MS for Bottom- and Middle-Up Characterization of Proteins
Identification, monitoring, and, more importantly, linkage of critical quality attributes (CQAs) in processing parameters in a biopharmaceutical product is required to ensure the quality and manufacturing consistency of the product, but also its safety and efficacy during clinical and commercial development. Recently, bottom- up and middle-up liquid chromatography–mass spectrometry (LC–MS) characterization strategies using immobilized enzyme reactors (IMERs) in combination with multidimensional liquid chromatography coupled with high-resolution MS (MDLC–HRMS), as well as sophisticated software solutions, have been added to the analytical toolbox. These strategies not only allow faster characterization of post-translational modifications (PTMs) present in biotherapeutic proteins but also have the potential to provide a fully automated and unified bottom-up, middle-up, and intact LC–MS characterization approach.
Biotherapeutics account for approximately one-half of the newly commercialized products; thus, they have become a rapidly growing portion of the total pharmaceutical market. Many biopharmaceuticals currently approved constitute single-target recombinant monoclonal antibodies (mAbs) for indications in oncology, anti-immunity, and chronic inflammatory applications (1). However, given the extreme complexity of disease mechanisms, many companies are also focusing on the development of multispecific biotherapeutics (next-generation biologics) that engage two or more protein targets (2). The competitive landscape for developing biopharmaceutical proteins and the complexity of these proteins call for the development of new, efficient, rapid, automated, and robust analytical methods throughout their entire development stage, from candidate screening to commercialization.
Companies developing and manufacturing biopharmaceuticals have adopted the main principles of quality by design (QbD). These principles require the definition of desired product performance, identification of critical quality attributes (CQAs), and an understanding of the impact of process product parameters on CQAs (3). CQAs are physical, chemical, biological, or microbiological characteristics from which an appropriate limit, range, or distribution should be defined to ensure the desired product quality (4). Recombinant mAbs produced in mammalian cells are large and heterogeneous proteins of approximately 150 kDa. Their heterogeneity results from the presence of a wide variety of post-translational modifications (PTMs), such as oxidation, deamidation, and glycosylation, that can occur during their manufacturing, storage, and post-administration (5). CQAs are identified by thorough characterization of PTMs using risk-filtering strategies and require specific ranges. Potential product quality differences resulting from manufacturing changes require a comprehensive characterization and evaluation of batch-to-batch variation of CQAs to ensure their safety, efficacy, stability, and batch-to-batch consistency (6).
The characterization of PTMs has been standardized and has mainly used variations of bottom-up or middle-up liquid chromatography–mass spectrometry (LC–MS) approaches. Bottom-up LC–MS approaches, commonly called peptide mapping, consist of identifying and quantifying PTMs at the peptide level after digesting the mAb with proteases such as trypsin, chymotrypsin, Lys-C, thermolysine, and Glu-C (7). Bottom-up LC–MS analyses are used to confirm the amino acid sequence as well as to identify, locate, and determine the relative amount of each modification while performing a thorough batch-to-batch comparison. Middle-up LC–MS refers to PTM analysis at the mAb subunit level (~25–100 kDa). Subunits are generated either by reducing the disulfide bonds to produce a light chain (LC), heavy chain (HC), or by digestion with proteases such as pepsin, papain, Lys-C, IgdE, and IdeS (8). In middle-up LC–MS analysis, the intact mass of the mAbs fragments is determined by providing information regarding the regional location of a modification or a sequence variant. This strategy has been used for differentiating Fc and Fab glycosylation and inter- and intra-free sulfhydryls analysis (9,10). Bottom-up and middle-up LC–MS are complementary and provide a powerful analytical tool for the comprehensive characterization of PTMs in mAbs.
The generation of samples for bottom-up or middle-up LC–MS analysis is typically performed manually and involves several tedious and labor-intensive steps. Recently, efforts have been concentrated in reducing analysis time and throughput by implementing sample preparation automation, enzyme immobilization, better instrumentation, faster and more sensitive mass spectrometers, and sophisticated software solutions.
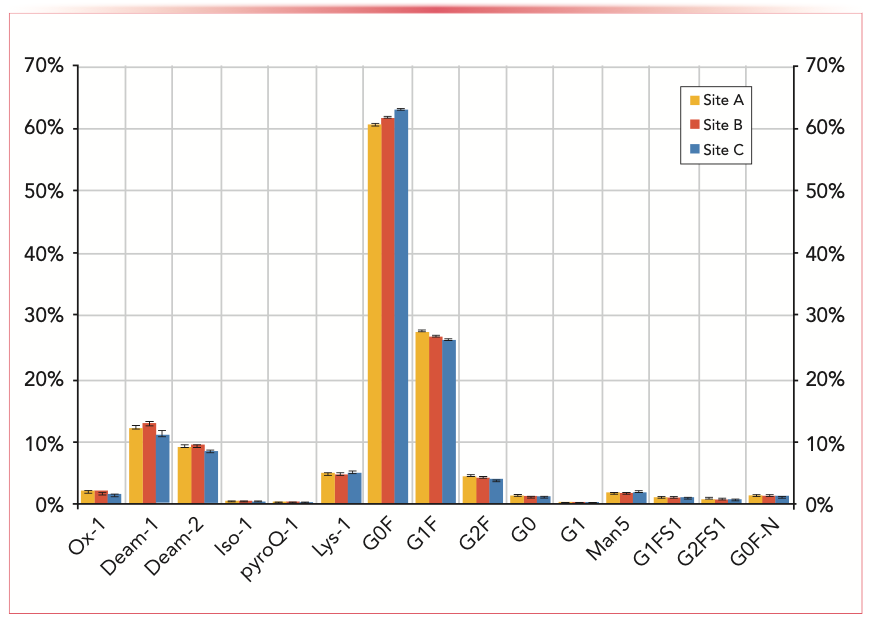
Bottom-up approaches, such as multi-attribute monitoring (MAM), benefit from the implementation of liquid handlers for sample preparation, as reported by Song and others (11). The application includes fully automated sample preparation using protein A purification and tryptic digestion, LC–MS-based MAM using full MS (MS1) data acquisition, and automated data analysis. This workflow supports cell line development, cell culture process, and downstream process development. Tryptic-digested samples were prepared in an automated fashion on a Hamilton Starline workstation, which was shown to increase throughput, productivity, accuracy, and precision. The harmonized digestion method was implemented across three different sites for monitoring CQAs. Robustness studies showed minimal quantification variability of CQAs, such as oxidation, deamidation, isoaspartic acid, C-terminal lysine, and major glycoforms (G0, G1, G0F, G1F, G2F, Man5, G1FS1, G2FS1, and GOF-N), over 96 injections of a single sample across sites (Figure 1).

 of the same antibody digest (mAb 1) analyzed at three different sites. Error bars indicate the standard deviation for each CQA over nine injections. The relative level of each CQA is based on the accumulated peak areas of all modified forms in relation to the total peak area of the corresponding CQA-carrying peptide (11).")
FIGURE 1: Comparison of CQA monitoring data of aliquots (n = 3) of the same antibody digest (mAb 1) analyzed at three different sites. Error bars indicate the standard deviation for each CQA over nine injections. The relative level of each CQA is based on the accumulated peak areas of all modified forms in relation to the total peak area of the corresponding CQA-carrying peptide (11).
Flow-through immobilized-enzyme reactors (IMERs) are enzyme-containing cartridges where an enzyme is covalently bound or immobilized on a solid support and packed into a column. This technique has been shown to maintain high enzyme specificity and catalytic efficiency, providing high accuracy and reproducibility (12). They have the advantage of operating in a continuous mode, which reduces analysis time (from hours to seconds) and cost, and also makes them amenable for automation. IMERs are used in many applications, such as drug metabolism research, enantioselective synthesis, identification of drug candidates, and novel inhibitors (13). In addition, IMERs provide comparable (and sometimes superior) performance compared to traditional offline digestion approaches, as described in the examples below.
IMERs have recently been used for analytical purposes such as offline, online, or multi-dimensional liquid chromatography–high-resolution mass spectrometry (MDLC–MS) analysis. Recent publications of online IMERs coupled to LC instruments have been used in the area of proteomics using enzymes such as Lys-C, Lys-N, pepsin, and PNGase-F (14). Perchepied and others reported the use of combined peptide mapping information from immobilized trypsin and pepsin for fast and reliable localization and characterization of N- and O-glycoforms in human chorionic gonadotropin hormone (HCG) by nano-LC–MS/MS and automated data analysis using Byonic (Protein Metrics). HCG is a highly glycosylated heterodimer protein with two N-glycosylation sites in the alpha and beta subunits and four O-glycosylation sites in the beta subunit. Performance of the combined trypsin and pepsin-IMER digestion results were compared to manual in-solution digestions. The combination of the two complementary IMER digestion modes resulted in the identification of 42 out of 45 common glycans in HCG using a digestion time of 30 min compared to overnight conventional sample preparation (15).
Similarly, Bonichon and others reported the development of a pepsin-IMER coupled to nano-LC–MS/MS for analysis and quantification of the human butyrylcholinesterase (HuBuChE) nonapeptide assay. This peptide is generated after digestion of HuBuChE that has reacted with organophosphorus compounds (OP), and it is a biomarker for nerve agent intoxication. Pepsin-IMER digestion parameters, such as digestion time, temperature, and amount of immobilized pepsin, were optimized. Digestion yields were monitored using a synthetic peptide as a calibrant by multiple reaction monitoring (MRM) analysis. Reproducibility was assessed by evaluating three pepsin-IMER preparations and the limit of quantification of the nonapeptide was estimated in the attomolar range. Digestion yields of HuBuChE were higher with the immobilized pepsin-IMER when compared to those generated by in-solution pepsin digestion (16).
Comprehensive characterization of glycan structure is important during the development of mAbs because their mechanism of action is mediated by the site and structure of the attached glycan. Sample preparation of the enzymatically released glycans is lengthy and requires sample clean-up and derivatization to facilitate their chromatographic separation and detection by LC–MS. Jmeian and others developed an online PNGase F-IMER to release neutral and acidic N-linked glycans from glycoproteins (RNAse B, Fetuin, A1AGP, IgG, thyroglobulin) and human blood serum coupled with LC–MS/MS analysis (17). The PNGase F-IMER was coupled to a C8 trap and porous graphitic carbon (PGC) HPLC-chip that allows fast and accurate glycan release, sample clean-up, LC separation, and MS detection. The method was able to detect released N-linked glycans from 100 fmol of protein and 0.1 μL of human blood serum at room temperature in 6 min reaction time. Neutral and acidic N-linked glycans were efficiently released from in-solution and human blood serum glycoproteins in a few minutes compared to overnight digestion at 37 °C, and the PNGase F-IMER was stable and able to operate continuously for over a month (17).
Further automation of sample analysis has been achieved by combining IMERS with specialized chromatographic techniques such as MDLC–HRMS. A fully automated middle-up LC–MS characterization of mAbs and bispecific antibodies (BsAbs) using an immobilized IdeS-enzyme HPLC column (Genovis) was reported by Camperi and others (8). The workflow consists of online digestion of untreated mAbs and BsAbs samples in the IdeS-HPLC column followed by the separation and reduction of the subunits by reversed-phase LC analysis and HRMS. Contrary to papain and pepsin, IdeS is highly specific and cleaves at the Gly236–Gly237 motif at the hinge region of the human IgG subclass 1–4, generating F(ab’)2 (≈100 kDa) and Fc (≈50 kDa) fragments. Additional reduction of these IdeS-generated fragments with either tris(2-carboxyethyl)phosphine (TCEP) or dithiothreitol (DTT) results in light chain, heavy chain, and Fd’ fragments of approximately 25 kDa each. The method was developed and implemented for the rapid characterization of PTMs as well as potential BsAbs product impurities because of chain pairing, which informed the process development of the molecule (8). This approach successfully streamlined extended characterization of mAbs with a much faster turnaround time compared to offline approaches.
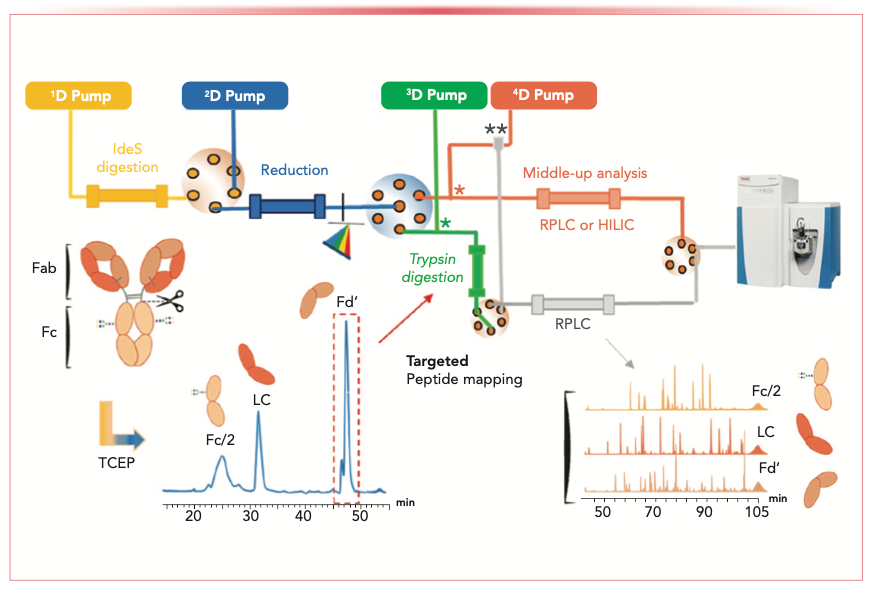
Applications were expanded to middle-up and targeted bottom-up 4D-LC–MS (digestion-IdeS ↔ reduction reversed-phase LC ↔ tryptic digestion ↔ reversed-phase LC–MS), and the workflow was reported for the comprehen- sive characterization of mAbs PTMs (18,19). Figure 2 shows the schematic of the setup. The workflow provides both reduced mass analysis information of the IdeS-generated fragments (light chain, heavy chain, and Fd’) and targeted peptide mapping data for each reversed-phase fraction, with complementary-determining region (CDR) sequence coverages higher than 97%. Comprehensive detection and quantification of multiple PTMs, such as oxidation, deamidation, and isomerization of each fraction, were demonstrated and compared to the offline method. In addition, the Fd’ fraction can be targeted for analysis of N-glycans, with a similar sensitivity to manual offline strategies (19).

, followed by on-column reduction by reversed-phase HPLC (2D, blue path), trypsin digestion in flow-through mode (3D, green path), and peptide mapping analysis by reversed-phase LC (4D, gray path) for each fragments Fd’, LC, and Fc/2. The selection of fragments is performed after the on-column reduction via a six-position, seven-port selector valve. Middle-up analysis using reversed-phase LC or HILIC for fragments separation (3D, orange path) can be performed within the same chromatographic system. The mixing tees and the zero dead-volume union were represented by (*) and (**), respectively (18,19).")
FIGURE 2: Schematic of the 4D-LC–MS workflow for a targeted peptide mapping approach using an online IdeS-HPLC column (1D, yellow path), followed by on-column reduction by reversed-phase HPLC (2D, blue path), trypsin digestion in flow-through mode (3D, green path), and peptide mapping analysis by reversed-phase LC (4D, gray path) for each fragments Fd’, LC, and Fc/2. The selection of fragments is performed after the on-column reduction via a six-position, seven-port selector valve. Middle-up analysis using reversed-phase LC or HILIC for fragments separation (3D, orange path) can be performed within the same chromatographic system. The mixing tees and the zero dead-volume union were represented by (*) and (**), respectively (18,19).
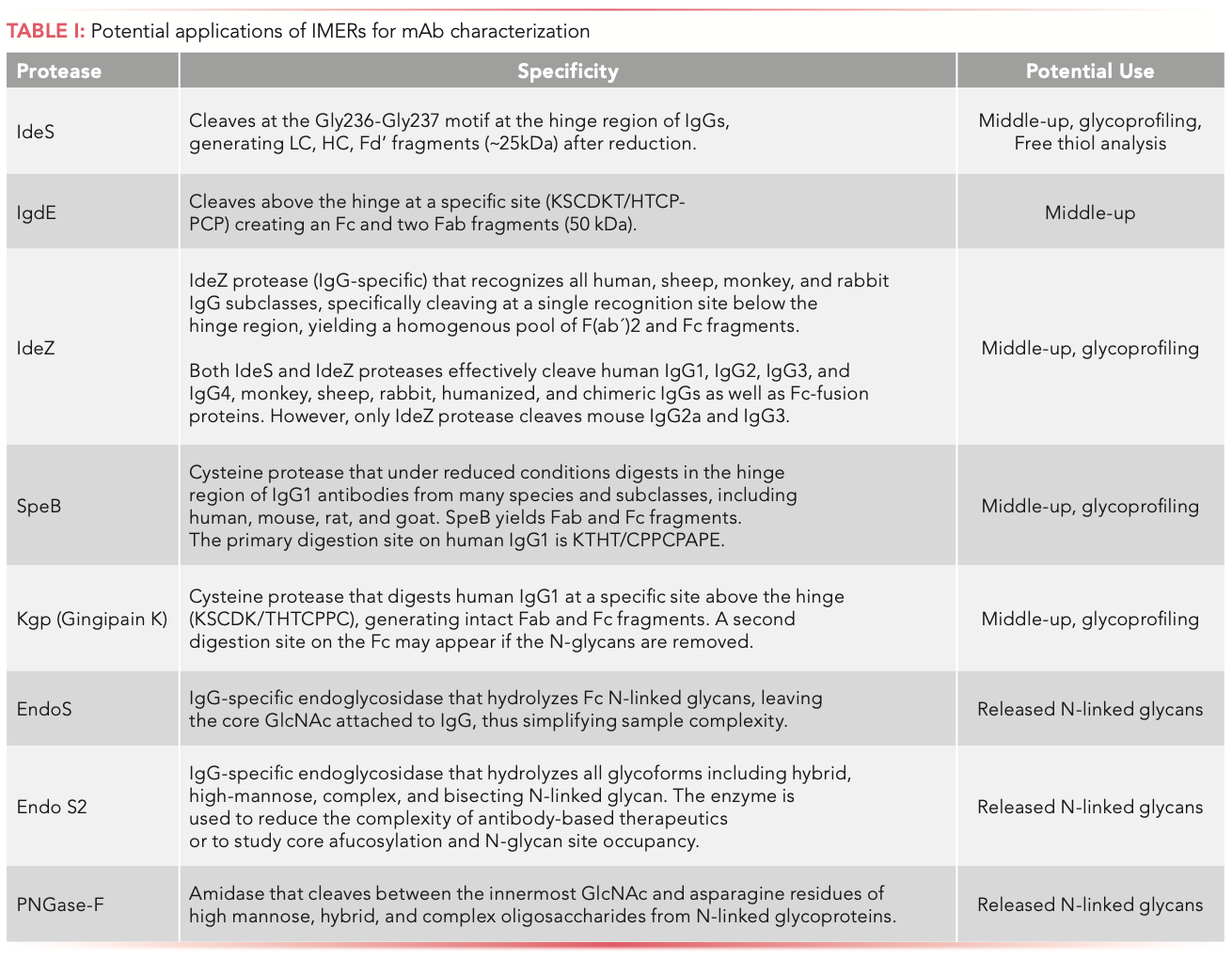
Key advantages of automation, enzyme immobilization (PNGase- F, IdeS, and trypsin), MDLC–HRMS instrumentation, and software solutions (not covered) are making a significant impact on the analysis of PTMs by allowing a more streamlined, yet still comprehensive identification, quantification, and monitoring. The implementation of these workflows can significantly reduce sample handling, operation time, and the risk of potential artifacts (oxidation and deamidation) compared to standard offline procedures. However, the required sophisticated instrumentation can be used to inform several stages of development but it may not yet be amenable for the quality control testing laboratories. In addition, this analytical workflow is extremely versatile as different types of immobilized enzymes (IdeZ, SpeB, IgdE, Kgp, Endo S2, Endo S) can be used, exponentially increasing the number of analytical protein applications to support current and novel biopharmaceutical formats (see Table I).


References
(1) D.E. Johnson, Int. J. Mol. Sci. 19, 11 (2018). DOI: 10.3390/ijms19113685.
(2) X. Zhong and A.M. D’Antona, Antibodies (Basel) 10, 2 (2021). DOI: 10.3390/antib10020013.
(3) A. Rathore and H. Winkle, Nat. Biotechnol. 27(1), 26–34 (2009). DOI: 10.1038/nbt0109-26.
(4) International Conference on Harmonization, ICH Q8(R2), Pharmaceutical Development (ICH, Geneva, Switzerland, 2009).
(5) X. Xu, et al, PLoS One 14(10), e0223899 (2019). DOI: 10.1371/journal.pone.0223899.
(6) O. Montacir, et al, J. Pharm. Biomed. Anal. 140, 239–251 (2017). DOI: 10.1016/j.jpba.2017.03.029.
(7) G. Pradhan, et al, J. Proteomics 232, 104053 (2021). DOI: 10.1016/j.jprot.2020.104053.
(8) J. Camperi, et al, J. Pharm. Biomed. Anal. 182, 113130 (2020). DOI: 10.1016/j.jpba.2020.113130.
(9) A.C. Robotham and J.F. Kelly, in Approaches to the Purification, Analysis and Characterization of Antibody-Based Therapeutics (Elsevier, Amsterdam, the Netherlands, 2020), pp. 1–33.
(10) V. Faid, et al, J. Pharm. Biomed. Anal. 149, 541–546 (2018). DOI: 10.1016/j.jpba.2017.11.046.
(11) Y.E. Song, et al, J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 1166, 122540 (2021). DOI: 10.1016/j.jchromb.2021.122540.
(12) C.L. Cardoso, et al, J. Chromatogr. A 1120(1–2), 151–157 (2006). DOI: 10.1016/j.chroma.2005.10.063.
(13) S.M. Fang, et al, J. Pharm. Anal. 2(2), 83–89 (2012). DOI: 10.1016/j.jpha.2011.12.002.
(14) B. Wouters, et al, TrAC Trends Anal Chem. 144, 116419 (2021). DOI: 10.1016/j.trac.2021.116419.
(15) S. Perchepied, et al, Talanta 206, 120171 (2020). DOI: 10.1016/j. talanta.2019.120171.
(16) M. Bonichon, et al, J. Chromatogr. A 1461, 84–91 (2016). DOI: 10.1016/j.chroma.2016.07.058.
(17) Y. Jmeian, L.A. Hammad, and Y. Mechref, Anal. Chem. 84(20), 8790– 8796 (2012). DOI: 10.1021/ac301855v.
(18) J. Camperi, et al, Anal. Chem. 92(6), 4357–4363 (2020). DOI: 10.1021/acs.analchem.9b05193.
(19) J. Camperi, D. Guillarme, and C. Stella, Anal. Chem. 92(19), 13420–13426 (2020). DOI: 10.1021/acs.analchem.0c02780.


Patricia Molina is a protein biochemist with broad experience in analytical characterization of therapeutic antibodies. Patricia has provided analytical method support for several pipeline molecules for the last seventeen years, including assay development, qualification, and validation. Patricia received her MS with an emphasis in biochemistry from San Francisco State University. She is currently a scientist at Genentech, Inc., a member of Roche Group in South San Francisco, California. Direct correspondence to: molina.patricia@gene.com.



Julien Camperi is Technical Development Scientist with the Cell Therapy Engineering & Development department of Genentech in South San Francisco, California. Direct correspondence to: camperi.julien@gene.com.



 Through the Use of Multi-Isocratic Elution Mode")
 Coupled to LC–MS/MS for Bottom- and Middle-Up Characterization of Proteins")
 Deployed in Online MDLC–MS Applications")











, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.




Common Challenges in Nitrosamine Analysis: An LCGC International Peer Exchange
April 15th 2025A recent roundtable discussion featuring Aloka Srinivasan of Raaha, Mayank Bhanti of the United States Pharmacopeia (USP), and Amber Burch of Purisys discussed the challenges surrounding nitrosamine analysis in pharmaceuticals.

