Advances in Hydrophilic Interaction Liquid Chromatography for Pharmaceutical Analysis
Special Issues
Hydrophilic interaction liquid chromatography (HILIC) has recently become more important, particularly for the analysis of polar drugs, metabolites and biologically relevant compounds in glycomics, proteomics, metabolomics and clinical analysis. HILIC makes it possible to increase the retention of polar compounds, achieve orthogonal selectivity and increase mass spectrometry (MS) sensitivity, compared with reversed-phase liquid chromatography. This article discusses the advantages and limitations of HILIC in a variety of practical applications in the pharmaceutical industry.
Hydrophilic interaction liquid chromatography (HILIC) has recently become more important, particularly for the analysis of polar drugs, metabolites and biologically relevant compounds in glycomics, proteomics, metabolomics and clinical analysis. HILIC makes it possible to increase the retention of polar compounds, achieve orthogonal selectivity and increase mass spectrometry (MS) sensitivity, compared with reversed-phase liquid chromatography. This article discusses the advantages and limitations of HILIC in a variety of practical applications in the pharmaceutical industry.
The term hydrophilic interation liquid chromatography (HILIC) was originally proposed by Alpert in 1990 (1) to describe an alternative strategy to normal-phase liquid chromatography (NPLC), ion-exchange (IEX) or ionpairing chromatography (IPC) for the analysis of polar substances, which are poorly retained in reversed-phasefffliquid chromatography. A large variety of biologically active substances including peptides, amino acids, carbohydrates, neurotransmitters, oligosaccharides, nucleotides or nucleosides and pharmaceutical compounds can be easily analysed with this approach. Apart from polar compounds, McCalley demonstrates that HILIC is also a viable strategy for analyzing a wide range of ionizable compounds (2).
Retention Mechanism and Stationary Phases for HILIC
HILIC is characterized by a hydrophilic stationary phase [including bare silica or silica derivatized with various polar functional groups such as amine, amide, cyano, diol etc (3)] with an aqueous organic solvent as the mobile phase. The latter should contain more than 60–70% of aprotic organic solvent, ideally acetonitrile. As can be seen from Figure 1, HILIC can be regarded as a kind of hybrid application between reversed-phase LC (similar mobile phase, organic modifier, buffer and pH range), normal-phase LC (similar stationary phases) and IEX (similar type of compounds that can be analysed).


Figure 1: Hydrophilic interaction liquid chromatography (HILIC) combines the characteristics of reversed-phase liquid chromatography (RPLC), normal-phase LC (NPLC) and ion-exchange chromatography (IEX). Adapted from reference (6).
Alongside the increasing popularity of HILIC, its interaction mechanism has received much attention over recent years and appears to be more complex than reversed-phase LC because of the relatively broad range of stationary phases that can be used in HILIC (3,4,5). It is now well established that a multimodal retention mechanism occurs, involving (i) the hydrophilic partitioning between a water-enriched layer at the stationary phase surface and the less polar eluent of the mobile phase, (ii) the ion-exchange between a charged analyte and the stationary phase, and (iii) the adsorption of the analyte at the surface of the adsorbent via hydrogen bonds. The extent of these different mechanisms strongly depends on the nature of the stationary phase, the type of organic solvent, the analysed compounds, the mobile phase pH and its ionic strength.

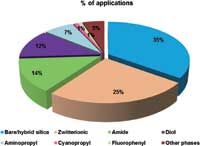
Figure 2: Polar bonded phases available for hydrophilic interaction liquid chromatography separations and their respective percentage of applications reported in scientific literature. Source: Scifinder scholar 2007 search of the Chemical Abstracts database from 2003 to 2012. Date of information gathering: December 2012.
A wide variety of packings can be used for HILIC experiments and the number of commercially available columns has continuously grown over recent years. However, no versatile stationary phase like C18 in reversed-phase LC is yet available (6). Among the polar chromatographic surfaces that can be used for HILIC separations, the unmodified bare or hybrid silica remain the most widespread and have been used in approximately 35% of the reported applications (Figure 2). These phases often provide suitable peak shape but the cation exchange mechanism plays a major role in retention and needs to be considered, especially for the analysis of positively charged compounds. The second most important type of bonding for HILIC is the zwitterionic sulphoalkylbetaine, which has been used in around 25% of the reported applications. The ligand at the surface of the phase contains both sulphonic acid (strong acid) and quaternary ammonium (strong base), separated by a short alkyl spacer.
This phase was originally intended for ion exchange separations but it has been successfully used for the simultaneous analysis of anionic and cationic compounds in HILIC (4). Amide, diol and aminopropyl also represent some promising stationary phases for HILIC and are usually prepared by simply modifying the silica gel surface. With amide phases (around 14% of the HILIC applications), the retention is not affected so much by ion exchange interactions and this type of bonding is particularly well suited for the separation of peptides, oligosaccharides and glycoproteins. Diol phases (approximately 12% of the HILIC applications) demonstrate high polarity and hydrogen bonding properties, such as bare silica, but do not contain ionizable groups other than residual silanols. Aminopropyl phases (less than 10% of the HILIC applications) are particularly relevant for the retention of acidic compounds via hydrophilic partitioning and ion exchange interactions. Indeed, this phase was found suitable for the analysis of sugars because it increases the rate of anomer mutarotation, which avoids the formation of double peaks.
Finally, there is a huge number of additional "exotic" phases that can be used in HILIC (less than 10% of the reported HILIC applications), including cyanopropyl, fluorophenyl, cyclodextrin, polysuccinimide, sugar phases and so on, but their use is limited to specific samples.
Advantages and Limitations of HILIC
The main advantage of HILIC is its ability to retain hydrophilic compounds without the need for toxic and expensive solvents such as those used in normal-phase LC, and without significant amount of salts or ion-pairing reagents, hardly compatible with MS detection. As well as polar compounds, HILIC can also be considered as a complementary technique to reversed-phase LC for the analysis of ionizable compounds such as drugs. Indeed, the elution order is almost reversed in HILIC (hydrophilic partitioning) compared to reversed-phase LC (hydrophobic partitioning). However, because of the strong contribution of an ion exchange mechanism, the achieved selectivity is generally very different in HILIC as opposed to reversed-phase LC, for example, in the case of peptides separation (7,8). Another major benefit of the HILIC mode is the enhanced signal in MS, explained by the more efficient desolvation of highly organic mobile phases (low density and surface tension of acetonitrile compared to water). A sensitivity gain of up to 10 times can be achieved in hydrophilic interaction liquid chromatography–electrospray ionization–mass spectrometry (HILIC–ESI–MS) compared with reversed-phase LC–ESI–MS (9), depending on the nature of the compounds, the mobile phase composition (10) and the ionization source geometry. As a result of a low viscosity of highly organic mobile phase (on average 2–3 times lower than reversed-phase LC), the generated backpressure is also limited, which allows higher flow rates with longer columns and smaller particle sizes to be used. This has been illustrated by McCalley (11) who reached efficiencies in excess of 100,000 plates within reasonable analysis times (less than 15 min) when using a 45-cm column.
In bioanalysis, solid-phase extraction (SPE) remains the primary sample preparation technique. However, the final elution step of SPE is usually performed with a large fraction of organic solvent that may be incompatible with the direct reversed-phase LC analysis. Therefore, a time-consuming evaporation or reconstitution step is generally added at the end of the extraction process. With HILIC separation, this highly organic eluent can be directly injected into the column, thus increasing the throughput (9). Finally, McCalley has also recently demonstrated that the loading properties of HILIC phases for basic solutes may be superior to those observed in reversed-phase LC because of the strong contribution of ion exchange mechanism (Figure 3) (12).

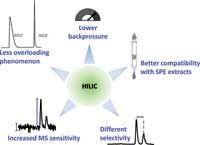
Figure 3: Main advantages of hydrophilic interaction liquid chromatography, compared to reversed-phase LC.
Despite this, HILIC is not as straightforward a technique as reversed-phase LC and there could be some reluctance to adopt such a technique even with its distinct advantages (13). The main drawback of HILIC is related to the availability and price of acetonitrile, which was a significant concern during times of shortage. Some attempts have been made recently to replace acetonitrile with acetone, but its applicability was rather limited with ultraviolet (UV) and MS detections. Since ion exchange is a kinetically slow process and strongly contributes to the interaction mechanism in HILIC, the equilibration time between successive gradient runs was also approximately two to four times longer in HILIC compared to reversed-phase LC. Nevertheless, no systematic studies have been conducted to precisely quantify this difference, which could be strongly dependent on the type of column in both techniques. An additional challenge of HILIC arises from the accurate measurement of mobile phase pH, as well as the analyte and silanols pKa, when using a large proportion of organic modifier within the mobile phase. Indeed, it is well established that pH and pKa values are affected by the presence of organic solvents (14), which makes it difficult to evaluate the ionization state of the analytes and the silanols, as well as the strength of ion exchange mechanism. For example,
McCalley estimated that an acetonitrile/ammonium formate buffer 85/15 (v/v), where the aqueous buffer pH was 3, presented an apparent pH of approximately 5.2 under HILIC conditions (2). Another common issue observed with HILIC is the peak shapes which may appear more distorted and broader than in reversed-phase liquid chromatography, especially for inexperienced users. This behaviour can be explained by the nature of the dissolution solvent which appears to be more critical in HILIC than in reversed-phase LC, as demonstrated in a recent systematic study (7). As reported, the sample diluent should be composed of the highest possible proportion of acetonitrile with a maximum of 10–20% water. Finally, the fact that the HILIC interaction mechanism (sum of hydrophilic partitioning + ion exchange + adsorption) is not as simple as in reversed-phase LC (which is mostly hydrophobic partitioning) is not an advantage. Indeed, the well-identified method development approaches of reversed-phase LC, which are highly valued and characterized, cannot be easily applied (13) and new strategies have to be developed.
New Trends in HILIC for Pharmaceutical Analysis
Column Technology for High Throughput HILIC Separations: Highthroughput separations are increasingly required for applications where the response time has to be reduced. With its need for enhanced productivity and reduced costs, the pharmaceutical field is considered to be the main driving force for faster separations. Because of the large number of analyses required for common pharmaceutical applications, such as, for example, purity assays, quality control and bioanalysis, including pharmacokinetic and drug metabolism studies; rapid analytical procedures (less than 5 min, including equilibration time) are usually required (15).
Over the last decade, various approaches have been developed to speed up reversed-phase LC analysis. Nowadays, two strategies are particularly interesting: (i) short columns packed with sub-2-µm fully porous particles under very high pressure, up to 1300 bar (UHPLC technology), and (ii) columns packed with sub-3-µm superficially porous particles, which are composed of a solid inner core of approximately 1.7-µm diameter surrounded by a thin layer of around 0.5-µm porous material (core–shell technology). With these two types of innovative supports, it is possible to drastically improve the throughput in comparison to the conventional 150 × 4.6 mm, 5 µm reversed-phase LC column, with a 6- to 9-fold reduction in analysis time for similar plate counts. The main drawback of these two approaches, and particularly with ultrahighperformance liquid chromatography (UHPLC), is the important pressure required for the percolation of the mobile phase through a bed of small particles. Hence, a dedicated instrument able to work beyond the 400-bar upper limit of conventional HPLC hardware is required. Moreover, the frictional heating effects induced by the elevated pressure drop produce axial and longitudinal temperature gradients inside the column, which may be detrimental to the separation quality (16).
Similar to reversed-phase LC, efficiency and column backpressure increase with the decrease of particle size in HILIC. However, working in HILIC with a proportion of acetonitrile comprised between 60% and 95% represents an obvious advantage, related to the low viscosity of the mobile phase (two to three times lower than in reversed-phase LC). Consequently, the reduced pressure drop under HILIC conditions allows the use of columns packed with sub-2-µm particles even on a classical HPLC instrument, provided that extra-column volume and dwell volume have been optimized for the column dimensions (17). Moreover, the frictional heating effect which may be detrimental in reversed-phase LC with sub-2-µm particles is no longer an issue because backpressure still remains reasonable in HILIC. As shown recently (18), the backpressure observed with a 100 × 2.1 mm HILIC column packed with sub-2-µm particles was equal to only 238 bar at 0.4 mL/min, which is approximately three times less than the same column dimension used under reversed-phase LC conditions at an equivalent flow rate. In a more systematic study (19), Chauve and colleagues compared the kinetic performance of several UHPLC and core–shell unmodified bare silica phases through van Deemter curves and kinetic plot representations. This work clearly demonstrated a significant gain in performance with these innovative technologies compared to conventional materials. An example is provided in Figure 4 and demonstrates the successful separation of various drugs and metabolites with fully porous sub2-µm (Waters Corporation) and core–shell sub3µm particles (Sigma Aldrich) in less than 4 min. However, because of the very low backpressure in HILIC, the characteristics of the core–shell column were not as attractive as in reversed-phase LC. Indeed, columns packed with sub-2-µm particles always provide better absolute kinetic performance in HILIC mode and should be selected for efficiency up to 200,000 plates.

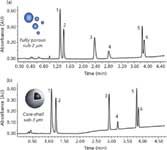
Figure 4: Separation of three drugs and their main metabolites with (a) column packed with fully porous sub-2-µm particles: 100 à 2.1 mm, 1.7 µm and (b) column packed with coreâshell sub-3-µm particles: 100 à 2.1 mm, 2.7 µm. Compounds: 1 = OH-midazolam; 2 = midazolam; 3 = bupropion; 4 = OH-bupropion; 5 = dextromethorphan; 6 = dextrorphan. See (19) for experimental conditions. Adapted and reprinted from J. Sep. Sci. 33(6â7), B. Chauve, D. Guillarme, P. Cléon and J.L. Veuthey, Evaluation of various HILIC materials for the fast separation of polar compounds, 752â764 (2010) with permission from Wiley-VCH Verlag GmbH (19)
A wide range of HILIC columns packed with fully porous sub-2-µm particles and superficially porous sub-3-µm particles are commercially available, including unmodified silica and hybrid silica, or silica bonded with amide, cyano, diol, zwitterionic or amino groups. The large choice of column chemistries and providers makes sub2-µm and core–shell HILIC phases particularly promising, even if they have only been used in approximately 10% of the reported HILIC applications up until now.
Automated Method Development Procedures in HILIC: Developing new analytical methods generally requires substantial time and effort, even for experienced chromatographers. In the pharmaceutical industry, the first step in a method development strategy often includes a generic trial-and-error screening protocol, to test several columns and mobile phase conditions in a systematic way and to find out the best combination of parameters for subsequent method optimization. In reversed-phase LC, these rules are well established and the best combinations that should be tested during this preliminary step have already been reported (20–22).
In HILIC, the stationary phase chemistry plays a major role and influences the compound retention and selectivity more than the mobile phase composition does. This observation has been made in several independent and recently published studies (4,23–29). Indeed, the understanding of the physico-chemical properties of a substance is essential to the selection of the suitable HILIC phase, assuming the significant diversity of available supports. Nevertheless, a limited number of stationary phases (four to five different chemistries) can be considered during the initial screening of unknown compounds. The choice of the useful phases may be determined from the following contributions (4,23–29): (i) unmodified bare or hybrid silica, even if a strong adsorption of many types of polar compounds has been reported (5); (ii) amide bonded phase, because of its high versatility for efficient and fast separation of polar drugs; (iii) amino phase because of the sufficient retention of acidic compounds; and (iv) zwitterionic sulphoalkylbetaine and poly(succinimide) bonded phases which also offer suitable retention for a wide range of analytes but with a lower kinetic performance compared to the other supports (4). Based on the availability of columns packed with sub-2-µm particles, Periat and colleagues (24) proposed an alternative set of four different HILIC columns for the initial screening: unmodified bare silica, unmodified hybrid silica, silica bonded with zwitterionic groups and hybrid silica bonded with amide. A set of model probes and principal component analysis (PCA) representation or radar plots have been employed in this study to characterize the type of interactions (hydrophilic, hydrophobic, electrostatic, hydrogen bonding, dipole-dipole, π-π interaction and shape-selectivity) that occur for a significant number of HILIC supports. Aside from the phase chemistry, the pH of the mobile phase was the most important parameter for tuning selectivity and retention, and it should also be considered in the initial generic screening protocol. Fountain and colleagues (23) proposed to test two different mobile phase pH — pH 3 and 9 — but only hybrid stationary phases can withstand such elevated pH. Since the number of hybrid HILIC phases is rather limited, Periat and colleagues (24) proposed to preferentially test pH 3 and 6 during the initial screening step.
For the screening procedure, less than 1 hr was required to test four different columns packed with sub-2-µm particles at the two pH (a total of eight conditions), using a generic gradient from 95% (1 min) to 65% acetonitrile in 3 min (with re-equilibration time of 3 min), on a 50 × 2.1 mm i.d. column at 500 µL/min. Depending on the achieved result, the separation could be further optimized by adjusting secondary parameters, such as, buffer ionic strength, gradient profile, nature or organic modifier or mobile phase temperature (24).
Application of HILIC in Pharmaceutical Analysis
Numerous pharmaceutical applications have been reported over the last decade with HILIC, including the analysis of drug compounds, their impurities and metabolites in either bulk material, pharmaceutical formulations or body fluids (blood, plasma, serum and urine). Excellent reviews describe these applications in detail (30–32). This section aims to highlight a few innovative approaches or remarkable results, and is not an exhaustive review of the reported applications.
Drug analysis with HILIC has been evaluated using a large set of pharmaceutical compounds covering a wide range of physico-chemical properties. In the first study, 45 drugs possessing log DpH3 between -3 and 4 were selected by Ruta et al. (18). Because most of the drugs were ionizable and presented a basic moiety, more than 90% were sufficiently retained under HILIC conditions. In addition, the achieved selectivity was found to be very different from that obtained in reversed-phase LC. In another work (24), an extended set of 82 drugs with log P values between -2 and 9 was investigated using numerous HILIC phases and mobile phase conditions. Again, the retention of drugs was satisfactory (around 90%) with various mobile and stationary phase conditions. Only the less polar acids were hardly retained because of electrostatic repulsions with the negatively charged surface of the HILIC phases. These two studies obviously confirm the applicability of HILIC in achieving orthogonal selectivity and retention compared to reversed-phase LC. HILIC was thus considered to analyse various types of polar active pharmaceutical ingredients (APIs) in formulations, as reported in (30). In addition, HILIC was also used in drug development for the separation of synthetic starting material, impurities, intermediates, excipients and APIs. Various examples are presented in reference (30). Interestingly, reversed-phase LC and HILIC columns were coupled in series to broaden the elution window for the simultaneous analysis of both polar and apolar drugs. This innovative method was applied for the concurrent analysis of sugars and sulphonamides as presented in Figure 5, or for genotoxic impurities, including arylamines and aminopyridines (33).


Figure 5: Separation of sugars and sulphonamides using (a) reversed-phase LC gradient analysis and (b) gradient reversed-phase LC/isocratic hydrophilic interaction liquid chromatography analysis. Compounds: 1 = ribose; 2 = glucose; 3 = sucrose; 4 = lactose; 5 = raffinose; 6 = sulphamerazine; 7 = sulphamethizole; 8 = sulphamethazine; 9 = sulphamethoxazole; 10 = sulphaquinoxaline. See (33) for experimental conditions. Adapted and reprinted from J. Chromatogr. A. 1208(1â2), S. Louw, A.S. Pereira, F. Lynen, M. Hanna-Brown and P. Sandra, High-efficiency hydrophilic interaction chromatography by coupling 25cmÃ.6mm IDõm silica columns and operation at 80 °C, 90â94 (2008) with permission from Elsevier B.V. (33).
Many lead optimization processes involve some bioanalytical assays to support both in vivo and in vitro experiments during drug discovery and development (32). Because of their inherent high sensitivity and selectivity, liquid chromatography–tandem mass spectrometry (LC–MS–MS) systems are currently considered the gold standard for the analysis of drugs in biological matrices. As mentioned previously, a higher sensitivity may be afforded by HILIC rather than conventional reversed-phase LC, because of an increased sensitivity in ESI provided by the highly volatile organic mobile phase. In addition, HILIC has proven to be a valuable technique for the analysis of polar drugs and metabolites; the latter being chemically more polar than their parent drug.
Finally, the organic extracts collected after protein precipitation (PP) or SPE are compatible with the higher organic content of HILIC mobile phases and can thus be directly injected onto a HILIC column without the need for time-consuming evaporation and reconstitution steps. Therefore, HILIC has rapidly gained in popularity in recent years for quantitative bioanalysis, with a predominant use of underivatized bare silica and zwitterionic sulphoalkylbetaine HILIC phases. Plasma and serum are the primary biological fluids handled in quantitative bioanalysis. However, as drugs and metabolites are mainly excreted via the kidneys, their quantitation in urine is required and can be highly informative about drugs exposure and distribution (31). Because of the inherent complexity of urine and the presence of large amounts of metabolic waste, biological salts and polar organic compounds, HILIC can be a useful approach for separating interfering compounds from the analytes of interest. Various applications of HILIC in urine analysis have been published and have shown a satisfying performance (31,32).
Matrix effect is one of the major concerns that can affect bioanalytical assay reliability. This phenomenon occurs when endogenous interferents co-elute with the analyte of interest and leads to signal suppression or enhancement during the ionization process (34).
According to the Food and Drug Administration (FDA) guidance, the matrix effect needs to be investigated as an integrated part of quantitative LC–MS–MS method development and validation procedure. Because of their high abundance in biological matrices, phospholipids are well known to be the major cause of matrix effects and are well retained by the HILIC phase. Therefore, matrix effects may be more critical in HILIC compared with reversed-phase LC. Phospholipids should thus be eliminated or at least reduced by using a suitable sample clean-up procedure or by adjusting the mobile phase pH, shifting the retention time of the target compounds without changing that of the phospholipids.
Finally, to achieve results of appropriate quality, a quantitative method needs to be fully validated to meet the strict regulatory requirements in the pharmaceutical analysis. This validation process has already been successfully performed in a HILIC quantitative method (30).
Conclusion
The exponentially growing number of applications on HILIC highlights the rising interest in this technique. HILIC presents some obvious advantages compared to reversed-phase LC in pharmaceutical analysis, such as (i) higher retention of polar drugs and metabolites, (ii) orthogonal or complementary selectivity to reversed-phase LC for complex mixture of pharmaceutical compounds, and (iii) improved MS sensitivity in bioanalytical applications. However, HILIC is still not as well illustrated as reversed-phase LC and some reluctance to adopt this technique may be observed in the industry.
Two important trends have emerged in pharmaceutical analysis to further improve the possibilities offered by HILIC. A large variety of HILIC columns packed with porous sub-2-µm and core–shell sub3µm particles are available, allowing a significant improvement of throughput and resolution. Secondly, generic screening protocols involving several well balanced stationary phases and mobile phase pH have been proposed for highly efficient method development, allowing for the best combination of stationary phase and mobile phase pH to be determined in less than an hour, thanks to columns packed with sub-2-µm particles.
References
(1) A.J. Alpert, J. Chromatogr. 499, 177–196 (1990).
(2) D.V. McCalley, J. Chromatogr. A 1171(1–2), 46–55 (2007).
(3) P. Hemström and K. Irgum, J. Sep. Sci. 29(12), 1784–1821 (2006).
(4) P. Jandera, Anal. Chim. Acta 692(1–2), 1–25 (2011).
(5) T. Ikegami, K. Tomomatsu, H. Takubo, K. Horie and N. Tanaka, J. Chromatogr. A 1184(1–2), 474–503 (2008).
(6) B. Buszewski and S. Noga, Anal. Bioanal. Chem. 402(1), 231–247 (2012).
(7) J. Ruta, S. Rudaz, D.V. McCalley, J.L. Veuthey and D. Guillarme, J. Chromatogr. A 1217(52), 8230–8240 (2010).
(8) M. Gilar, P. Olivova, A.E. Daly and J.C. Gebler, Anal. Chem. 77(19), 6426–6434 (2005).
(9) E.S. Grumbach, D.M. Wagrowski-Diehl, J.R. Mazzeo, B. Alden and P.C. Iraneta, LCGC North America 22, 1010–1023 (2004).
(10) H.P. Nguyen and K.A. Schug, J. Sep. Sci. 31(9), 1465–1480 (2008).
(11) D.V. McCalley, J. Chromatogr. A 1193(1–2), 85–91 (2008).
(12) D.V. McCalley, J. Chromatogr. A 1217(6), 858–880 (2010).
(13) J. Heaton and N.W. Smith, Chromatography Today 5(2), 44–47 (2012).
(14) S. Espinosa, E. Bosch and M. Roses, Anal. Chem. 72(21), 5193–5200 (2000).
(15) D. Guillarme, J. Ruta, S. Rudaz and J.L. Veuthey, Anal. Bioanal. Chem. 397(3), 1069–1082 (2010).
(16) L. Novakova, J.L. Veuthey and D. Guillarme, J. Chromatogr. A 1218(44), 7971–7981 (2011).
(17) S. Fekete and J. Fekete, J. Chromatogr. A 1218(31), 5286–5291 (2011).
(18) J. Ruta, J. Boccard, D. Cabooter, S. Rudaz, G. Desmet, J.L. Veuthey and D. Guillarme, J. of Pharm. Biomed. Anal.63, 95–105 (2012).
(19) B. Chauve, D. Guillarme, P. Cléon and J.L. Veuthey, J. Sep. Sci. 33(6–7), 752–764 (2010).
(20) L.R. Snyder, J. Chromatogr. B Biomed. Sci. Appl 689(1), 105–115 (1997).
(21) I. Krull, M. Schwartz, J. Turpin, P.H. Lukulay and R. Verseput, A Quality-byDesign Methodology for Rapid LC Method Development, Part I,LCGC North America (December 2008).
(22) T. Baczek, Current Pharm. Anal. 4(3), 151–161 (2008).
(23) K.J. Fountain, J. Xu, D.M. Diehl and D. Morisson, J. Sep. Sci. 33(6–7), 740–751 (2010).
(24) A. Periat, B. Debrus, S. Rudaz and D. Guillarme, J. Chromatogr. A1282, 72–83 (2013).
(25) R.I. Chirita, C. West, A.L. Finaru and C. Elfakir, J. Chromatogr. A1217(18), 3091–3104 (2010).
(26) M. Lämmerhofer, M. Richter, J. Wu, R. Nogueira, W. Bicker and W. Lindner, J. Sep. Sci. 31(14), 2572–2588 (2008).
(27) Y. Guo and P.G. Wang, in W. He (Ed.), Hydrophilic Interaction Chromatography (HILIC) and Advanced Applications, (CRC Press, New York, USA 2011), p. 401.
(28) N.P. Dinh, T. Jonsson and K. Irgum, J. Chromatogr. A 1218(35), 5880–5891 (2011).
(29) Y. Guo and S. Gaiki, J. Chromatogr. A 1218(35), 5920–5938 (2011).
(30) B. Dejaegher and Y. Vander Heyden, J. Sep. Sci. 33(6–7), 698–715 (2010).
(31) W. Jian, R.W. Edom, Y. Xu and N. Weng, J. Sep. Sci. 33(6–7), 681–697 (2010).
(32) Y. Hsieh, J. Sep. Sci.31(9), 1481–1491 (2008).
(33) S. Louw, A.S. Pereira, F. Lynen, M. HannaBrown and P. Sandra, J. Chromatogr. A 1208(1–2), 90–94 (2008).
(34) I. Marchi, V. Viette, F. Badoud, M. Fathi, M. Saugy, S. Rudaz and J.L. Veuthey, J.Chromatogr. A 1217(25), 4071–4078 (2010).
Aurélie Périat is a Ph.D. student at the School of Pharmaceutical Sciences at the University of Geneva in Switzerland. She is working on the possibilities and limitations of HILIC and HILIC–MS for pharmaceutical analysis.
Isabelle Kohler is a Ph.D. student at the School of Pharmaceutical Sciences at the University of Geneva, Switzerland. Her main work focuses on the hyphenation of CE with MS in clinical and forensic toxicology, and she is involved in the investigation of improved sample preparation techniques in bioanalysis (microextraction techniques) prior to LC–MS or CE–MS analysis.
Davy Guillarme holds a Ph.D. degree in analytical chemistry from the University of Lyon, France. Since 2004, he has been a senior lecturer at the University of Geneva in Switzerland. His expertise includes HPLC, UHPLC, HILIC, LC–MS, analysis of proteins and mAbs and, more recently, SFC.
Jean-Luc Veuthey obtained his Ph.D. from the University of Geneva, Switzerland in 1987. He is now a full professor in the Department of Pharmaceutical Sciences, University of Geneva and University of Lausanne, Switzerland. His interests include the development of LC and CE hyphenated to several detection modes for the analysis of drugs and metabolites.



















