Adjustment to the Chromatographic System in U.S. Pharmacopeia Monographs: Flexibility and Limitations
LCGC North America


To meet system suitability requirements, chromatographers might need to make adjustments to the high performance liquid chromatography (HPLC) operating conditions.
The description of a chromatographic procedure in a US Pharmacopeia (USP) monograph contains necessary information that enables the procedure to be reproduced in a laboratory and typically includes system suitability requirements. System suitability is based upon the concept that "the equipment, electronics, analytical operations, and samples to be analyzed constitute an integral system that can be evaluated as such" (1), and it is used to demonstrate the system is stable and suitable for its intended use. In some instances, the system fails to meet those requirements, resulting in the need for corrective actions. The deviations can be temporary (short-term excursions caused by a particular factor or combinations of factors) or more permanent. The causes for such deviations might be assignable or might be of uncertain origin attributable to normal system variability.



Because this situation can arise, the System Suitability section of USP's General Chapter Chromatography <621> acknowledges that adjustments to operating conditions might be necessary to meet system suitability requirements and includes the maximum variation allowed for each chromatographic parameter. Users are allowed to implement these adjustments to bring the system into suitable performance without a full validation of the analytical procedure. The influence of the changes should be assessed by verification of the procedure under the adjusted chromatographic conditions. Chromatographers must demonstrate that the changes introduced will not affect the performance of the procedure adversely. If the allowed adjustments to the system are not successful and assistance from USP staff has not resolved the problem, the procedure might no longer be suitable for use with the article being tested. This conclusion could be a laboratory-specific issue (for example, a bad column) or it could be serious enough to warrant revision of the existing monograph. This article discusses the flexibility and limitations of the System Suitability section in the current version of <621> and provides some specific examples to help analysts understand how to take advantage of the flexibility that is allowed in this chapter.
Adjustments to the Chromatographic System
As mentioned, <621> allows a number of adjustments to the chromatographic system in response to issues with a procedure's system suitability. These are the maximum allowable variations unless the individual monograph directs otherwise.
Ratio of components in mobile phase: Although the replacement of any solvent in the mobile phase constitutes a change and not an adjustment, the amounts of the minor components (specified as 50% or less) in the mobile phase can be adjusted up to ±30% relative to the particular component. However, the change in any component cannot exceed ±10% absolute (that is, in relation to the total mobile phase). Adjustments can be made to one minor component in a ternary mixture.
pH of mobile phase: The pH of the aqueous buffer used in the preparation of the mobile phase can be adjusted to within ±0.2 units of the value or range specified.
Column temperature: The column temperature can be adjusted by as much as ±10 °C.
Concentration of salts in buffer: The concentration of the salts used in the preparation of the aqueous buffer for the mobile phase can be adjusted up to ±10% relative to the particular component, provided the permitted pH variation (see above) is met.
Wavelength of UV–vis detector: Deviations from the wavelengths specified in the method are not permitted.
Stationary phase:
- Column length: The column length can be adjusted by as much as ±70%.
- Column inner diameter: The column inner diameter can be adjusted, provided that the linear velocity is kept constant (see Flow Rate).
Flow rate: When column dimensions have been modified, the flow rate can be adjusted using the following formula:

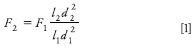
where
F1 is the flow rate indicated in the monograph, in milliliters per minute;
F2 is the adjusted flow rate, in milliliters per minute;
l1 is the length of the column indicated in the monograph;
l2 is the length of the column used;
d1 is the column inner diameter indicated in the monograph; and
d2 is the internal diameter of the column used.
Additionally, the flow rate can be adjusted by ±50% (2).
Injection volume: The injection volume can be reduced as far as is consistent with accepted precision and detection limits, but no increase is permitted.
Particle size: Particle size can be reduced by as much as 50% but cannot be increased.
The degree of verification needed to implement these adjustments varies and depends upon the purpose of the procedure, the extent of the changes, and the performance characteristics that could be affected by the changes — see USP General Information Chapter Verification of Compendial Procedures <1226> (3). Meeting system suitability requirements might not be sufficient to demonstrate that the procedure has been verified successfully. Especially in older monographs, the requirements for the system suitability test might be weak, and other experiments might be needed to demonstrate acceptable system performance.
The following examples demonstrate the flexibility and limitations offered by the current version of <621>.
Mobile Phase Composition
Mobile phase composition is one of the primary causes for the inability to meet the system suitability criteria provided in an official monograph or in a monograph proposal published in Pharmacopeial Forum (PF). An example is the olanzapine proposal that appeared in PF 34(3) (4). The proposal included the following chromatographic conditions and system suitability criteria:
Mobile phase: Buffer–acetonitrile (50:50).
Resolution requirement: Not less than (NLT) 2.0 between olanzapine-related compound A and olanzapine.
During evaluation of the PF proposal, some users observed that the resolution requirement could not be met easily. Figure 1 shows the chromatogram obtained under the conditions in the PF proposal.

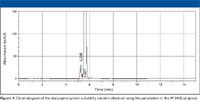
Figure 1
The observed resolution was 1.7. According to <621> for the specified ratio of 50:50, 30% percent of 50 is 15% absolute, but this exceeds the maximum permitted change of ±10% absolute in either component. Therefore, the mobile phase ratio of buffer–acetonitrile can be adjusted only within the range of 40:60 to 60:40. Decreasing the organic component relative to the buffer is potentially a way to obtain the required resolution. In this case, when the organic content of the mobile phase composition was changed to 47% (buffer–acetonitrile 53:47), the resolution improved to 7.0 (Figure 2).


Figure 2
USP received a comment from another user stating that the drug substance has a specified process impurity that elutes very close to the drug substance. Lowering the mobile phase organic component is likely to improve the separation between the drug substance and the process impurity as well. Presented with this information, the USP Expert Committee responsible for the approval of this monograph decided to revise the mobile phase composition to buffer–acetonitrile (53:47) in the official monograph. Changing only the mobile composition according to <621> might not lead to successful results in all cases. Thus, the chapter recommends that users contact USP staff for assistance. The next example demonstrates the outcome of an interaction between the user and USP staff.
The paroxetine hydrochloride monograph in USP 29 (5) had the following chromatographic conditions:
- Mobile phase composition: acetate buffer–acetonitrile–triethylamine (60:40:1).
- System suitability requirements: resolution between paroxetine-related compound B and paroxetine NLT 2.0.
- Tailing factor requirement for paroxetine peak: Not more than (NMT) 1.6.
Users reported that after one week of use with a new column, neither the tailing factor nor the resolution requirement could be met with Zorbax TMS, the column used in the validation studies. Although the mobile phase includes three components, lowering the acetonitrile content is likely to result in higher resolution. A systematic evaluation showed that 70% aqueous buffer (30% acetonitrile) was necessary to achieve the required resolution.
The new chromatographic conditions included the following:
- Mobile Phase: acetate buffer–acetonitrile–triethylamine (70:30:1).
- Column: 25 cm × 4.6 mm Zorbax TMS
- Temperature: 30 °C
- Flow rate: 1.0 mL/min
- Observed resolution: 2.5
- Tailing factor: 1.8.
The new mobile phase conditions resulted in a tailing factor of more than 1.6. Tailing factors obtained in a robustness study with perturbation of column temperature and mobile composition were all greater than 1.6 (Tables I and II, respectively).

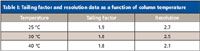
Table I: Tailing factor and resolution data as a function of column temperature
Because the tailing factor did not exceed 2.0 under any of the chromatographic conditions used in the robustness study, it was concluded that a tailing factor requirement of NMT 2.0 would be acceptable. The monograph was revised as follows (6):

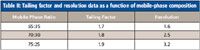
Table II: Tailing factor and resolution data as a function of mobile-phase composition
Mobile phase: acetate buffer, acetonitrile, and triethylamine (70:30:1). (Note: The actetate buffer–acetonitrile–triethylamine ratio can be varied between 70:40:1 and 75:25:1 to meet the system suitability requirements.)
The recommended range of variation (70:40:1 and 75:25:1) for the mobile phase composition included in the paroxetine hydrochloride monograph was outside the range allowed in <621>, so this was included in the text of the monograph (6). The tailing factor requirement was increased from NMT 1.6 to NMT 2.0.
Column Temperature
A proposal submitted for the escitalopram tablets monograph included a related compounds test using a column temperature of 50 °C, and no range was indicated. This means that the column temperature can vary between 40 and 60 °C in accordance with the variation allowed in <621>. Robustness data showed that the resolution between the critical pair was sensitive to temperature and that resolution was lost when a temperature of 55 °C was used. Because the acceptable adjustment to the temperature range is relatively tight compared with the allowances in <621>, the escitalopram tablets monograph (7) specifies a temperature range of 50 × 2 °C.
pH of the Mobile Phase
The proposal for escitalopram tablets monograph included a mobile phase pH requirement of 7.0 with a resolution requirement between the critical pair of 1.2. This means that the pH of the mobile phase can range between 6.8 and 7.2 according to the allowed variation in <621>. Robustness data (Table III) showed that resolution between the critical pair was sensitive to pH, and the resolution decreased as the pH of the mobile phase increased. The monograph for escitalopram tablets (7) specifies a pH range of 7.0 × 0.1.

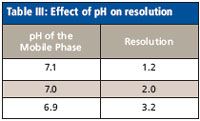
Table III: Effect of pH on resolution
Column Dimensions: Inner Diameter, Length
The chromatographic purity test included in the hydroxyzine hydrochloride monograph (8) required the use of two 10 cm × 3 mm columns coupled in series and containing packing L3 with a flow rate of 0.4 mL/min. It would be advantageous to use one column with standard dimensions of 4.6 mm i.d. and a 25-cm length. Chapter <621> allows the simultaneous change of column inner diameter and length and recommends the flow rate be adjusted according to equation 1 given earlier.
The new flow rate calculated using the formula is 1.2 mL/min. When a flow rate of 1.0 mL/min was used with a 4.6-mm i.d. and 25-cm length column, the observed resolution was greater than 3. The current official monograph includes the revised column dimensions of 4.6-mm i.d. and 25-cm length and flow rate of 1.0 mL/min (9).
Injection Volume
A user-performed verification studies for the paroxetine hydrochloride monograph (5) according to <1226> and found that the spike recovery of paroxetine-related compound C was consistently low (Table IV).


Table IV: Spike recovery of paroxetine-related compound C using 1.0 mg/mL standard solution
The procedure is used to monitor 0.1% (w/w) of paroxetine-related compound C (1 mg/mL). The procedure uses 5 μL of a paroxetine-related compound C solution with a concentration of 1 mg/mL, so the amount of paroxetine-related compound C injected on column is 5 μg. The concentration of paroxetine in the sample solution is 5 mg/mL, which translates to 0.025 mg of paroxetine-related compound C at 0.1% (w/w), which is 200 times lower than the absolute amount of the standard. Chapter <621> allows lowering the sample injection volume, but the injection volume of 5 μL is low. Lowering the injection volume by a factor of 10 to 0.5 μL is not feasible using standard HPLC equipment, and the injection precision requirement of not more than 2.0% relative standard deviation (RSD) likely would not be met. The other option is to lower the concentration of the standard solution to 0.1 mg/mL and maintain the 5-μL injection volume. Because this is not an option in <621>, validation of the procedure is necessary to assess the impact of such a change. Table V shows the observed spike recovery with the 0.1 mg/mL standard solution. Spike recoveries given in Table V are indicative of normal variability.
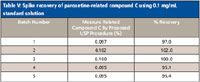
Table V: Spike recovery of paroxetine-related compound C using 0.1 mg/mL standard solution
The injection precision requirement of NMT 2.0% RSD could not be met because the concentration of the standard was lowered. The monograph was revised to allow the use of lower standard concentration (0.1 mg/mL) and a higher RSD of not more than 10.0%, which is supported by the validation data (6).
Multiple Adjustments — Column Particle Size and Mobile Phase Component
The clotrimazole monograph uses an HPLC procedure for the assay as well as for detection and quantitation of impurities (9). The official monograph includes the following chromatographic conditions:
- Mobile phase: methanol–buffer (75:25)
- Column: 25 cm × 4.6 mm, 10 μm
- Flow rate: 1.5 mL/min
- System suitability requirements: Resolution between clotiramzole and clotiramzole-related compound A NLT 1.9.
A user reported that the resolution requirement could not be met using the mobile phase conditions provided in the monograph even with a new column. Following changes permitted by <621>, the mobile phase composition was modified to methanol–buffer (65:35). Although the resulting resolution of 2.2 met the system suitability requirement, the baseline was unstable and a notable reduction in the response of clotrimazole was observed. No assignable cause was found for this observation. The resolution requirement also could have been met by using a 5-μm column, but that would not have addressed the issues regarding baseline noise and loss of signal. Use of acetonitrile instead of methanol and a 5-μm column solved the unstable baseline problem and restored the response. Although the change from a 10-μm to a 5-μm particle was within the guidelines, changing the major solvent (for example, methanol to acetonitrile) is more than an adjustment and therefore full validation of the new method would be required. Chapter <621> states "Multiple adjustments can have a cumulative effect in the performance of the system and should be considered carefully before implementation." In this case, the multiple changes, including the change of a major solvent component, were considered extensive and, thus, required the user to validate the new chromatographic procedure.
Conclusions and Future Trends
The system suitability section of <621> provides flexibility in a number of chromatographic system parameters. Although the recommended ranges included in this chapter may be useful in achieving the system suitability requirements specified in the monograph, in some situations, the flexibility provided might not resolve the issues. In such situations, users should work with USP staff to evaluate the need to revise the monograph in question.
USP's General Chapters Expert Committee has been exploring ways to increase the flexibility allowed in USP monograph testing. A potential revision to Chapter <621> suggests a process that will allow analysts, without extensive revalidation, to use HPLC columns with dimensions and particle sizes that differ from those prescribed in the monograph. This concept is developed further in a stimuli article that will appear in (11).
Acknowledgements
The authors would like to thank Teva Industries and Sun Pharmaceuticals for allowing the use of their data in this article.
Dr. Ravi Ravichandran has been with USP since 2004. He is the scientific liaison to both the Expert Committee on Psychiatric and Psychoactives and the Expert Committee on Radiopharmaceuticals and Medical Imaging Agents. Ravi received his degree in Chemistry from Annamalai University in India and earned a Ph.D. in Analytical Chemistry from University of Louisville, Louisville, KY and post-doc with the late Prof. L.B. Rogers, University of Georgia.
Dr. Horacio N. Pappa has been with USP since 2003. He is the scientific liaison to both the Expert Committee on General Chapters and the Expert Committee on Statistics. Horacio received his degree in Pharmacy as well as a Ph.D. in Pharmaceutical Chemistry at Buenos Aires University.


Ronald E. Majors
Ronald E. Majors "Column Watch" Editor Ronald E. Majors is Senior Scientist, Columns and Supplie Division, Life Sciences Chemical Analysis, Agilent Technologies Wilmington, Delaware, and is a member of LCGC's editorial advisory board. Direct correspondence about this column to "Column Watch," LCGC, Woodbridge Corporate Plaza, 485Route 1 South, Building F, First Floor, Iselin,NJ 08830, e-mail lcgcedit@lcgcmag.com.
References
(1) USP. USP 32–NF 27, Chromatography <621>. Rockville, MD: USP; 2009:227.
(2) USP. Second Supplement to USP 32–NF 27. Rockville, MD: USP; 2009:4147.
(3) USP. USP 32–NF 27, Verification of Compendial Procedures <1226>. Rockville, MD: USP; 2009:736.
(4) USP. Pharm Forum. Olanzapine Monograph. 2008;34(3):641.
(5) USP. USP 29–NF 24. Rockville, MD: USP; 2006:1644.
(6) USP. USP 32–NF 27, Paroxetine Hydrochloride Monograph. Rockville, MD: USP; 2009:3214–3216.
(7) USP. Authorized USP Pending Standards: Escitalopram Tablets. 2009 http://www.usp.org/pdf/EN/pendingStandards/escitalopramTabletsAuthorized.pdf This monograph was developed according to USP's Pending Standards policy that can be found at http://www.usp.org/standards/pending/guidelines.html
(8) USP. USP 29–NF 24, Hydroxyzine Hydrochloride Monograph. Rockville, MD: USP; 2009:1089–1090.
(9) USP. USP 32–NF 27, Hydroxyzine Hydrochloride Monograph. Rockville, MD: USP; 2009:2596–2597.
(10) USP. USP 32–NF 27, Clotrimazole Monograph. Rockville, MD: USP; 2009:1997–1998.
(11) U.D. Neue, D. McCabe, V. Ramesh, H. Pappa, and J. DeMuth, Pharm Forum., in press.













. | Image Credit: © Joan Vadell - stock.adobe.com")
Detecting Hyper-Fast Chromatographic Peaks Using Ion Mobility Spectrometry
May 6th 2025Ion mobility spectrometers can detect trace compounds quickly, though they can face various issues with detecting certain peaks. University of Hannover scientists created a new system for resolving hyper-fast gas chromatography (GC) peaks.

Altering Capillary Gas Chromatography Systems Using Silicon Pneumatic Microvalves
May 5th 2025Many multi-column gas chromatography systems use two-position multi-port switching valves, which can suffer from delays in valve switching. Shimadzu researchers aimed to create a new sampling and switching module for these systems.





