Validation of LC–MS-MS Methods for the Determination of Ibuprofen in Miniature Swine Plasma and Synovial Fluid
Special Issues
Published methods for the determination of ibuprofen in biological fluids by liquid chromatography (LC)–UV or LC–mass spectrometry (MS)-MS have quantitation ranges consistent with the relatively high but typical ibuprofen dose (200–800 mg), generally having lower limits of quantitation in the low micrograms-per-milliliter range. For the analysis of plasma and synovial fluid samples from preclinical (miniature swine) studies utilizing a novel ibuprofen dosage form, LC–MS-MS methods were developed and validated over the 10–1000 ng/mL range. Ibuprofen undergoes biotransformation to ibuprofen acyl glucuronide and sublimes under routine bioanalytical sample handling conditions. Procedures were implemented to minimize the impact of these potential liabilities.
Published methods for the determination of ibuprofen in biological fluids by liquid chromatography (LC)–UV or LC–mass spectrometry (MS)-MS have quantitation ranges consistent with the relatively high but typical ibuprofen dose (200–800 mg), generally having lower limits of quantitation in the low micrograms-per-milliliter range. For the analysis of plasma and synovial fluid samples from preclinical (miniature swine) studies utilizing a novel ibuprofen dosage form, LC–MS-MS methods were developed and validated over the 10–1000 ng/mL range. Ibuprofen undergoes biotransformation to ibuprofen acyl glucuronide and sublimes under routine bioanalytical sample handling conditions. Procedures were implemented to minimize the impact of these potential liabilities.
Ibuprofen (Figure 1) is a nonsteroidal anti-inflammatory drug (NSAID) of the 2-arylpropionic acid class indicated in the treatment of rheumatoid and osteoarthritis and other inflammatory conditions including bursitis, tendonitis, low-back pain, and soft-tissue injuries such as sprains and strains. It also is indicated as an antipyretic and analgesic for the relief of mild to moderate pain (1). The anti-inflammatory effects of ibuprofen are linked to its inhibition of the arachidonate cyclooxygenase (COX) enzymes, with desirable effects resulting from the inhibition of COX-2 and adverse effects (nausea, dyspepsia, gastrointestinal ulceration or bleeding, raised liver enzymes, diarrhea, constipation, and so forth) primarily resulting from the inhibition of COX-1 (2).


Figure 1: Structure of ibuprofen.
Ibuprofen is metabolized extensively by means of formation of the major metabolites 2-hydoxyibuprofen and carboxyibuprofen. Both 3-hydroxyibuprofen and 1-hydroxyibuprofen have been detected in urine at low levels (3,4). All hydroxy and carboxy metabolites are pharmacologically inactive. Ibuprofen and its metabolites also undergo conjugation with glucuronic acid at the carboxylic acid moiety of the propionic side chain to yield acyl glucuronides (5).
Acyl glucuronides have reactive properties that stem from the electrophilic nature of the ester carbon atom, which can be subject to nucleophilic substitution reactions. At physiological pH, acyl glucuronides may hydrolyze, isomerize (by acyl migration), and covalently bind with macromolecules (proteins, DNA), the latter implicated in the toxicity of carboxylic acid drugs (6–8). Hydrolysis of acyl glucuronides is a hydroxide-initiated reaction resulting in the release of the original aglycone. Acidification of the matrix at the point of sample collection and during processing are procedural precautions often used for the stabilization of acyl glucuronides in bioanalytical methods, as degradation may lead to erroneously high levels of the parent drug.
To eliminate many of the adverse effects associated with ibuprofen therapy, a novel dosage form has been developed and tested in a miniature swine model. Both plasma and synovial fluid samples were collected, synovial fluid being the most relevant biological fluid surrogate for determining the therapeutic effect of anti-arthritic drugs. Synovial fluid is secreted by synovial cells lining the joint capsule, providing nutrients and lubrication for articular cartilage. The subsynovial layer contains lymphatics and blood vessels; consequently, blood contamination because of trauma is relatively common for synovial fluid samples obtained by means of joint aspiration (arthrocentesis). Synovial fluid contains a significant amount of hyaluronic acid (300 mg/dL), a high molecular weight polymerized glycosaminoglycan, which is responsible for the unique egg-white consistency and high viscosity of synovial fluid (9).
Published methods for the determination of ibuprofen in plasma and synovial fluid by high performance liquid chromatography (HPLC)–UV, gas chromatography–mass spectrometry (GC–MS), and LC–MS-MS (5,10,11) have quantitation ranges consistent with the relatively high but typical ibuprofen dose (200–800 mg), generally having lower limits of quantitation (LLOQ) in the low micrograms-per-milliliter range. For the determination of ibuprofen in plasma and synovial fluid samples from a miniature swine study utilizing a novel dosage form, relatively small sample volumes and an LLOQ in the low nanograms-per-milliliter range were required. To meet these requirements, LC–MS-MS methods were developed and validated using solid-phase extraction (SPE) and supported-liquid extraction (SLE) sample preparation procedures for plasma and synovial fluid, respectively.
Experimental
Chemicals and Reagents: Ibuprofen (sodium salt) was purchased from Sigma-Aldrich (St. Louis, Missouri). Ibuprofen-D3 internal standard was purchased from Toronto Research Chemicals (North York, Ontario, Canada) and ibuprofen acyl glucuronide was purchased from TLC PharmaChem (Vaughan, Ontario, Canada). Phosphoric acid and ammonium hydroxide were purchased from JT Baker (Phillipsburg, New Jersey), formic acid and dimethyl sulfoxide were purchased from Sigma-Aldrich, and acetic acid was purchased from EMD (Gibbstown, New Jersey). Blank miniature swine plasma collected from whole blood with K2EDTA anticoagulant and blank miniature swine synovial fluid were obtained from Sinclair Bio-resources (Columbia, Missouri). All solvents were HPLC grade and were purchased from VWR International (West Chester, Pennsylvania).
Stock and Spiking Solutions: Before weighing, ibuprofen was dried overnight at 104 °C to remove residual moisture. Ibuprofen stock solutions (1 mg/mL, corrected for purity and salt form) were prepared in duplicate in 50:50 (v/v) water–methanol and verified for accuracy of preparation before use. An ibuprofen-D3 stock solution (1 mg/mL) was prepared in dimethyl sulfoxide. All stock solutions were stored at –20 °C. Ibuprofen spiking solutions were prepared from a stock solution by serial dilution, with 50:50:0.1 (v/v/v) water–acetonitrile–formic acid as diluent. Spiking solutions were stored at 2–8 °C. All solutions were prepared fresh daily or used within established stability time-frames.
Calibration Standards and Quality Control Samples: Calibration standards and quality control (QC) samples were prepared by adding 50 µL of spiking solution to 950 µL of blank matrix, scaling up as required. Calibration standard concentrations were 10, 25, 50, 75, 100, 250, 750, and 1000 ng/mL; QC sample concentrations were 10, 30, 350, and 700 ng/mL. Calibration standards and QC samples were stored at ≤ –70 °C. For the assessment of dilution integrity, a dilution QC sample was prepared at 10,000 ng/mL and diluted 50-fold with blank matrix to be within the quantitation range (200 ng/mL).
SPE Procedure (Plasma): Spiked plasma (100 µL) was diluted in a 96-well plate with 100 µL of a working solution comprising 250 ng/mL ibuprofen-D3 in 2% phosphoric acid. Samples then were transferred to a 96-well Oasis HLB µElution plate (Waters Corp., Milford, Massachusetts) that had been conditioned with 200 µL of methanol and equilibrated with 200 µL of water. Samples were washed with 100 µL of 80:20:0.1 (v/v/v) water–acetonitrile–formic acid and eluted with 50 µL of 20:80:0.1 (v/v/v) water–acetonitrile–formic acid. Samples were diluted with 150 µL of 60:40:0.1 water–acetonitrile–formic acid.
SLE Procedure (Synovial Fluid): Blank synovial fluid was diluted threefold with water before spiking to reduce viscosity. Spiked synovial fluid (50 µL) was diluted in a microcentrifuge tube with 150 µL of pH 3.5 water (pH adjusted with formic acid) containing 250 ng/mL ibuprofen-D3. Samples were vortexed and centrifuged. Supernatants were transferred to a 96 well Isolute SLE+ supported liquid extraction plate (Biotage, Charlotte, North Carolina). Samples were drawn onto the extraction plate solid support using a vacuum manifold and extracted using 1 mL of a 90:10 (v/v) solution of dichloromethane–isopropanol, following the instructions provided with the plate. The extraction solvent was collected in a deep-well collection plate and reduced to dryness under nitrogen in a TurboVap 96 evaporator (Biotage). The samples were reconstituted in 75 µL of 60:40:0.1 (v/v/v) water–acetonitrile–formic acid and transferred to a shallow-well plate for injection.
LC–MS-MS Analysis: The LC–MS-MS system comprised a Shimadzu Prominence 20AD HPLC system (Shimadzu Scientific Instruments, Columbia, Maryland) with a 50 mm × 2.1 mm, 3.5-µm ZORBAX Eclipse XDB-C18 analytical column (VWR International) and an AB Sciex API 5000 mass spectrometer (AB Sciex, Foster City, California) operated in selected reaction monitoring (SRM) mode with negative electrospray ionization. The ibuprofen and ibuprofen-D3 SRM transitions were m/z 205→161 and m/z 208→164, respectively. Mobile phase A was 0.005% formic acid in water and mobile B was acetonitrile; the flow rate was 0.750 mL/min and the column temperature was 40 °C. Initial chromatographic conditions were 30% B with a 0.5 min hold followed by a linear gradient to 55% B at 4.50 min. A high organic (90% B) column wash and reequilibration to initial conditions completed the run cycle. The autosampler temperature was 15 °C and the injection volume was 10 µL. The retention time for ibuprofen and ibuprofen-D3 was 3.9 min and the total run time was 6.5 min. A basic autosampler wash, 50:50 (v/v) pH 8 ammonium hydroxide–methanol, was used to reduce carryover.
Assay Validation and Acceptance Criteria: Validation assessments were performed as per an in-house standard operating procedure, a document based on the 2001 FDA Guidance for Industry: Bioanalytical Method Validation (12) and current industry best practices (13). The following assessments were performed: selectivity, matrix effects, calibration curve, LLOQ, intra- and interassay accuracy and precision, carryover, recovery, dilution integrity, reinjection reproducibility, effect of hemolysis (plasma), blood contamination (synovial fluid), and interference from degradation of ibuprofen acyl glucuronide. Additionally, the following stability assessments were performed: stock, working and spiking solution stability, freeze–thaw stability, short-term stability, postpreparative stability, and long-term storage stability.
Results and Discussion
During the initial method development phase, both SLE and SPE were evaluated for plasma and synovial fluid sample preparation. For plasma, SPE had the benefits of a cleaner baseline and the use of solvents that would not require evaporation to dryness and reconstitution. Ibuprofen sublimation has been well-documented as an enthalpy-driven process resulting from unusual thermodynamic properties of the molecule. Sublimation has been reported at conditions of pharmaceutical relevance, for example, conditions used for drying or coating processes and conditions used for accelerated stability testing (15,16). The use of an SPE plate eliminated the need for sample dry-down and reconstitution, thereby eliminating any potential losses in recovery because of sublimation. Of the SPE sorbents available, both a neutral phase and anion-exchange phase were considered. Although an anion-exchange phase potentially could add additional selectivity, thereby producing an overall cleaner extract, elution from such a phase would require basic conditions, potentially degrading the acyl glucuronide metabolite. Using the neutral (HLB) phase, the SPE wash and elution steps were optimized to maximize recovery.
Because of its viscosity, synovial fluid is a challenging matrix. Dilution and warming eases sample handling; however, for the analysis of samples anticipated to have low ibuprofen levels, substantial dilution would be detrimental to the study, producing a significant number of results <LLOQ. For the method described herein, blank synovial fluid matrix was diluted threefold with water before spiking. The same procedure would be used for study samples, resulting in an effective LLOQ threefold higher than that represented by the lowest standard of the curve. An evaluation of SPE as a sample preparation procedure showed that synovial fluid samples would not flow freely through the wells of SPE plates, even after the primary threefold dilution and secondary dilution resulting from the addition of internal standard solution. In this regard, SLE was superior and selected for synovial fluid sample preparation.


Figure 2: Representative chromatograms from blank and spiked plasma: (a) double blank, (b) ibuprofen spiked at LLOQ, and (c) ibuprofen spiked at ULOQ.
Blank miniature swine plasma and synovial fluid from six different animals were used to evaluate selectivity. Samples were prepared that demonstrated the absence of significant interference from endogenous compounds, carryover, ibuprofen and the internal standard (cross-interference), and the sample preparation procedure, including the addition of glacial acetic acid at the point of sample collection as a stabilizer. A representative double-blank plasma chromatogram is shown in Figure 2a.

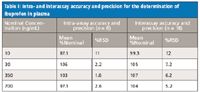
Table I: Intra- and interassay accuracy and precision for the determination of ibuprofen in plasma
Calibration standards were prepared in duplicate and run as bracketing curves at the beginning and end of each validation run. A 1/x weighting factor was applied and the curve was linear (r 2 > 0.999) over the calibration range (10–1000 ng/mL). All curve acceptance criteria were met throughout the validation exercise. Representative chromatograms at the lower and upper limits of quantitation (LLOQ and ULOQ, respectively) are shown as Figures 2b and 2c, respectively. Quality control samples (n = 6 at 10 [LLOQ], 30, 350, and 700 ng/mL) were prepared on each of three days for the assessment of intra- and interassay accuracy and precision. The results are summarized in Tables I and II for plasma and synovial fluid, respectively. Dilution integrity was evaluated by preparing a dilution QC sample (n = 6 at 10,000 ng/mL) and diluting 50-fold with blank matrix; all QC acceptance criteria were met.

Table II: Intra- and interday accuracy and precision for the determination of ibuprofen in synovial fluid
Ibuprofen acyl glucuronide is known to be labile, degrading by base-induced hydrolysis to the parent drug. The sample collection and preparation procedures utilized acidic conditions to prevent hydrolysis; nevertheless, potential interference from acyl glucuronide degradation was assessed. Quality control samples (n = 6 at 30 and 700 ng/mL) were prepared in the presence of 500 ng/mL ibuprofen acyl glucuronide, a concentration in excess of the expected physiological levels (14). All QC acceptance criteria were met in the presence of the metabolite, indicating the absence of significant metabolite degradation (Tables III and IV). The effects of hemolysis (for plasma) and blood contamination (for synovial fluid) were assessed by preparing QC samples (n = 6 at 30 and 700 ng/mL) in blank matrix containing 5% (by volume) hemolyzed whole blood. All QC acceptance criteria were met (Tables III and IV).

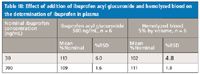
Table III: Effect of addition of ibuprofen acyl glucuronide and hemolyzed blood on the determination of ibuprofen in plasma
Extraction recovery and matrix effect were evaluated for both ibuprofen and ibuprofen-D3 at three QC levels (n = 6 at 30, 350, and 700 ng/mL). Extraction recovery was evaluated by comparing the results from pre-extraction spiked matrix to postextraction spiked matrix. The recovery from plasma (54–60%) and synovial fluid (37–41%) were consistent for both ibuprofen and the internal standard across the QC range. Matrix effect, quantified as the internal-standard normalized matrix factor, was evaluated by comparing results from postextraction spiked matrix to samples prepared in neat solution. Internal-standard normalized matrix factors of 0.99 and 1.0 were obtained for plasma and synovial fluid, respectively.
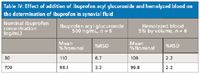
Table IV: Effect of addition of ibuprofen acyl glucuronide and hemolyzed blood on the determination of ibuprofen in synovial fluid
Ibuprofen and ibubrofen-D3 stock solution stability was established for 5 and 7 days, respectively, when stored at –20 °C. Ibuprofen working and spiking solution stability was established for 24 h when stored at ambient temperature and 4 °C. Ibuprofen-D3 working solution stability was established for 24 h when stored at ambient temperature.
All stability assessments with spiked plasma and synovial fluid samples were performed at two QC levels (n = 6 at 30 and 700 ng/mL). Short-term stability was established for 24 h at ambient temperature. Postpreparative stability was established for prepared samples stored at 4 °C for 24 h. Reinjection reproducibility was demonstrated by reinjecting previously injected samples after storage in the autosampler for 24 h at 15 °C. Freeze–thaw stability was established for three freeze–thaw cycles, where freezing (≤ –70 °C) was for a minimum of 12 h following an initial freezing of over 24 h, with room temperature thawing for a minimum of 4 h. Long-term storage stability was demonstrated for storage at ≤ –70 °C at periodic intervals up to six months.
Conclusions
Bioanalytical methods were developed and validated for the determination of ibuprofen in plasma and synovial fluid to support a miniature swine study utilizing a novel ibuprofen dosage form. Ibuprofen undergoes biotransformation to ibuprofen acyl glucuronide and sublimes under routine bioanalytical sample handling conditions. Procedures were implemented to minimize the impact of these potential liabilities.
The following validation assessments were performed: selectivity, matrix effect, calibration curve, LLOQ, intra- and inter-assay accuracy and precision, carryover, recovery, dilution integrity, re-injection reproducibility, effect of hemolysis (plasma), blood contamination (synovial fluid), and interference from degradation of ibuprofen acyl glucuronide. In all cases, results met acceptance criteria as per the 2001 Guidance (12). The validated methods were applied successfully to plasma and synovial fluid samples from a miniature swine study utilizing a novel ibuprofen dosage form.
Lawrence Andrade, Adam Grenier, Amber Awad, and Teresa Pekol are with Smithers Pharma Services, LLC, Wareham, Massachusetts.
References
(1) DrugBank, www.drugbank.ca.
(2) M. Kerola, K. Vuolteenaho, O. Kosonen, H. Kankaanranta, S. Sarna, and E. Moilanen, Basic & Clin Pharm & Tox. 104, 17–21 (2009).
(3) C. Giachetti, G. Zanolo, and S. Canali, J. High Res Chromatogr Commun. 8, 465–468 (1985).
(4) C.J.W. Brooks and M.T. Gilbert, J. Chromatogr. 99, 541–551 (1974).
(5) N. Davies, Clin Pharmacokinet. 34, 101–154 (1998).
(6) M. Shipkova, V. William Armstrong, M. Oellerich, and E. Wieland, Therapeutic Drug Monitoring 25, 1–16 (2003).
(7) S. Bolze, O. Lacombe, G. Durand, P. Chaimbault, F. Massiere, C. Gay-Feutry, N. Bromet, and T. Hulot, Current Separations 20, 55–59 (2002).
(8) M. Castillo and P.C. Smith, Drug Metab Dispos. 23, 566–572 (1995).
(9) A. Faryna, K. Goldenberg, Clinical Methods: The History, Physical, and Laboratory Examinations., H.K. Walker, W.D. Hall, J.W. Hurst, Ed. 3rd edition. (Boston, Butterworths, 1990). Chapter 166.
(10) V. Hynninen, K.T. Olkkola, K. Leino, S. Lundgren, P.J. Neuvonen, A. Rane, M. Valtonen, H. Vyyrylainen, and K. Laine, Antimicrob Agents Chemother. 50, 1967–1972 (2006).
(11) P.S. Bonato, M.P. Del Lama, and R. de Carvalho, J. Chromatogr B 796, 413–420 (2003).
(12) Food and Drug Administration. Guidance for Industry: Bioanalytical Method Validation. Rockville, Maryland: US Department of Health and Human Services, FDA, Center for Drug Evaluation and Research (2001).
(13) C.T. Viswanathan, S. Bansal, B. Booth, A.J. DeStefano, M.J. Rose, J. Sailstad, V.P. Shah, J.P. Skelly, P.G. Swann, and R. Weiner, The AAPS Journal 9, E30–E42 (2007).
(14) M. Castillo, Y.W. Lam, M.A. Dooley, E. Stahl, P.C. Smith, Clin Pharmacol Ther. 57, 636–644 (1995).
(15) S. Lerdkanchanaporn and D. Dollimore, J. Therm Anal. 49, 879–886 (1997).
(16) K.D. Ertel, R.A. Heasley, C. Koegel, A Chakrabarti, and J.T. Carstensen, J. Pharm Sci. 79, 552 (1990).










; An Interview with Fabrice Gritti")
: An Interview With Fabrice Gritti")


. From industry he went on to Florida State University, where he was assistant professor of both analytical and materials chemistry. Since 2011, he has been at the National Institute of Standards and Technology (NIST), where he is currently Scientific Advisor in the Chemical Sciences Division. André is the author of over 90 peer-reviewed scientific publications, lead author of the second edition of “Modern size-exclusion liquid chromatography,” editor of the book “Multiple detection in size-exclusion chromatography,” past associate editor of the Encyclopedia of Analytical Chemistry and, since 2015, editor of Chromatographia. He has received a number of awards, including the inaugural ACS-DAC Award for Young Investigators in Separation Science, and was also inaugural Professor in Residence for Preservation Research and Testing at the US Library of Congress. His interests lie principally in the area of macromolecular separations, both fundamental and applied.")


Determining the Effects of ‘Quantitative Marinating’ on Crayfish Meat with HS-GC-IMS
April 30th 2025A novel method called quantitative marinating (QM) was developed to reduce industrial waste during the processing of crayfish meat, with the taste, flavor, and aroma of crayfish meat processed by various techniques investigated. Headspace-gas chromatography-ion mobility spectrometry (HS-GC-IMS) was used to determine volatile compounds of meat examined.

, also known as an immunoglobulin (Ig). 3d vector © sakurra - stock.adobe.com")
Accelerating Monoclonal Antibody Quality Control: The Role of LC–MS in Upstream Bioprocessing
This study highlights the promising potential of LC–MS as a powerful tool for mAb quality control within the context of upstream processing.

